
We are excited to share GPN-Star, a cost-effective, biologically grounded genomic language modeling framework that achieves state-of-the-art performance across a wide range of variant effect prediction tasks relevant to human genetics.
www.biorxiv.org/content/10.1...
(1/n)
22.09.2025 05:29 — 👍 174 🔁 90 💬 4 📌 5
I am thrilled to announce that in January 2026 I will be starting my own lab at NYU Biology! Soon enough I will be recruiting postdocs and students! Please reach out if you are interested with a CV and description of your research interests, or if you know of people who could be interested! 🧬🗽 🦊
25.06.2025 20:10 — 👍 82 🔁 19 💬 7 📌 0
How can one efficiently simulate phylodynamics for populations with billions of individuals, as is typical in many applications, e.g., viral evolution and cancer genomics? In this work with M. Celentano, @wsdewitt.github.io , & S. Prillo, we provide a solution. doi.org/10.1073/pnas...
1/n
23.05.2025 21:02 — 👍 37 🔁 15 💬 1 📌 1

Thrilled to see my digital art on the cover of Trends Genet. The two binary strings represent reverse-complementary DNA sequences (00=A, 01=C, 10=G, 11=T) and the connecting rectangles represent “embeddings” learned by DNA language models. Pls check out our article as well: doi.org/10.1016/j.ti...
07.04.2025 15:01 — 👍 69 🔁 13 💬 0 📌 1
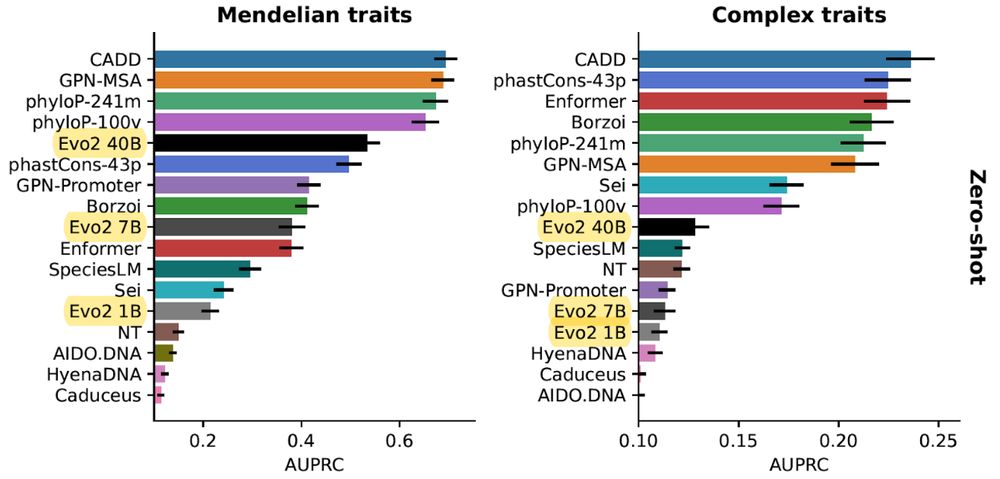
In our updated TraitGym preprint (w/ @gonzalobenegas.bsky.social & Gökcen Eraslan), we evaluate Evo 2 on regulatory variants associated with human traits. We see marked performance gains with scale on Mendelian traits, although still a bit behind alignment-based methods.
doi.org/10.1101/2025...
1/n
04.03.2025 19:54 — 👍 32 🔁 13 💬 1 📌 2
Thank you for contributing to bioicons! Sorry I forgot to add to acknowledgements, I will in the final version!
15.02.2025 19:29 — 👍 1 🔁 0 💬 0 📌 0
Thank you Remi!
14.02.2025 18:34 — 👍 0 🔁 0 💬 0 📌 0
Scaling is probably part of the solution, but data curation might be the major bottleneck. The vast majority of bases in mammalian genomes lack evolutionary constraint which is precisely the signal leveraged by self-supervision.
13.02.2025 20:57 — 👍 0 🔁 0 💬 1 📌 0
Alignment-free DNA language models are not yet competitive. The best among them, our GPN-Promoter and SpeciesLM from @gagneurlab.bsky.social , are not the largest in number of parameters or context. Their key feature is having been trained only on functional regions of the genome.
13.02.2025 20:57 — 👍 1 🔁 0 💬 1 📌 0

Conservation-aware CADD and GPN-MSA do better on Mendelian trait variants, expected to be under strong purifying selection. On complex trait variants, especially for non-disease traits, functional-genomics models Enformer and Borzoi tend to do better. However, ensembling helps:
13.02.2025 20:57 — 👍 1 🔁 0 💬 1 📌 0

We evaluate models zero-shot (unsupervised) and with linear probing (logistic regression on top of extracted features):
13.02.2025 20:57 — 👍 0 🔁 0 💬 1 📌 0

We evaluate a wide range of models with up to 7B parameters and 500K context size. Do these numbers matter? 🤔
13.02.2025 20:57 — 👍 0 🔁 0 💬 1 📌 0

We collect putative causal variants from OMIM and UKBB with carefully matched controls.
13.02.2025 20:57 — 👍 0 🔁 0 💬 1 📌 0
Can DNA sequence models predict mutations affecting human traits?
We introduce TraitGym, a curated benchmark of causal regulatory variants for 113 Mendelian & 83 complex traits, and evaluate functional genomics and DNA language models. Joint work w/ Gökcen Eraslan and @yun-s-song.bsky.social 🧵👇
13.02.2025 20:57 — 👍 28 🔁 15 💬 1 📌 2
Benchmarking DNA Sequence Models for Causal Regulatory Variant Prediction in Human Genetics https://www.biorxiv.org/content/10.1101/2025.02.11.637758v1
13.02.2025 07:33 — 👍 8 🔁 1 💬 0 📌 1
I still believe in alignment-free gLMs with better data curation and loss functions, I've been seeing advances but still tough.
02.02.2025 19:36 — 👍 1 🔁 0 💬 0 📌 0
*An exception are alignment-based gLMs which do improve (non-trivially) over conservation scores.
02.02.2025 19:36 — 👍 0 🔁 0 💬 1 📌 0
A simple bar is: do you surpass conservation scores in identifying functional mutations? This bar was easily passed by pLMs and plant gLMs but not yet by human gLMs* even after 5 years.
02.02.2025 19:35 — 👍 2 🔁 0 💬 1 📌 0
Thank you Jo!
12.01.2025 19:34 — 👍 0 🔁 0 💬 0 📌 0
Coincidentally, another article from my lab on DNA language models got published on the same day as GPN-MSA. It's freely available for 50 days from this link:
authors.elsevier.com/a/1kNCscQbJB...
Genomic language models: opportunities and challenges
Please share with your colleagues.
03.01.2025 02:29 — 👍 10 🔁 2 💬 1 📌 0
Thanks! Do you think this could explain part of the gap between task 4 and 5? Could profile prediction help generalization to variants (of the count head)?
11.12.2024 18:37 — 👍 0 🔁 0 💬 1 📌 0
Really appreciate your paper. Could you clarify about whether ChromBPNet and DNALMs are trained on the same data? From the paper and code it might seem like only ChromBPNet is given profile-level labels.
11.12.2024 03:09 — 👍 0 🔁 0 💬 1 📌 0

Ultrafast classical phylogenetic method beats large protein...
Amino acid substitution rate matrices are fundamental to statistical phylogenetics and evolutionary biology. Estimating them typically requires reconstructed trees for massive amounts of aligned...
Large protein language models can learn complex epistatic interactions, but how much does that help with predicting variant effects? In this NeurIPS article, we show that classical independent-sites phylogenetic models can outperform pLMs on this task.
1/7
openreview.net/forum?id=H7m...
16.11.2024 20:41 — 👍 91 🔁 44 💬 2 📌 2
Researcher @bayraktar_lab @teichlab @steglelab.bsky.social @sangerinstitute.bsky.social | Using models & AI to study cells, cell circuits & brains 🧠 | #SingleCell+spatial | 🌍+🇺🇦
Complex traits, gene regulation, ELSI (ethical, legal, and social implications of) genetics
PhD candidate with the Pritchard Lab @Stanford
Scientist and medical doctor. Biology AI/ML methods, gene regulation, DNA sequence models, single cells. Doing a PhD in computational biology at @molgen.mpg.de.
Incoming Assistant Professor @ MIT Biology, Koch Institute, and Institute for Medical Engineering and Science. Single-cell advisor to Vevo Therapeutics. Lineage tracing & cancer evolution. Website: thejoneslaboratory.com
bioinformatics phd student at UCLA
Data Scientist Generative AI @BayerCropScience. ML for Plant Biology. PhD @IowaStateUniversity https://www.linkedin.com/in/koushik-nagasubramanian/
Research scientist at @GoogleDeepMind passionate about AI, genomics and biology.
assistant prof at University of Oregon. interested in pop gen, stat gen, human complex traits. also ELSI, metascience, ethics education, etc...
she/they. 🌈
roshnipatel.github.io
Group Leader @ BIH Berlin and MPI Molecular Genetics. I play around with DNA because I‘d like to know how the genome functions.
🧬Genetics geek, 🤖AI enthusiast. PI @GenomeDataLab studying DNA repair, mutations, epigenome & NMD. 2xERC, professor @bric-ucph.bsky.social, GL @irbbarcelona.org. 🎲Chaotic good.
Ph.D. Student at Columbia University Biomedical Informatics | Machine Learning for Genomics.
Aspiring scientific educator with a love for writing and parks.
Research in AI for Protein Design @Harvard | Prev. CS PhD @UniofOxford, Maths & Physics @Polytechnique
Math Professor - IFCE
PhD Bioinformatics - UFMG
🧬 Human Genetics scientist | computational biologist | foodie | coffee snob ☕️ |🖖🏽🔬👨🏾💻🏳️🌈| all opinions here are my own 🐒
Exploring efficient computation, statistical inference, evolution, genetics & immunology 💻🧬📈. Postdoc, Barton Lab @ Pitt Comp & Sys Bio. PhD (Weigt Lab), Sorbonne Univ (UPMC).
PhD student in evolutionary biology @UChicago
Spatial & temporal structure of biodiversity and morpho complexity; evolution and genetics of complex traits
























