
We show that vasculature loss precedes muscle wasting in cancer cachexia. Years of work summed up in this tweetorial.
Also includes high quality muscle single cell data via GEO.
We show that vasculature loss precedes muscle wasting in cancer cachexia. Years of work summed up in this tweetorial.
Also includes high quality muscle single cell data via GEO.
There are three primary ways to use SenePy: 1) as an input list in any tool that takes a gene list, such as gene set enrichment. 2) senescence scoring directly on single-cell data. 3) Database to search for senescence markers in a specific cell type. (5/5)
24.02.2025 19:31 — 👍 0 🔁 0 💬 0 📌 0
In addition to aging, SenePy is widely applicable in many disease contexts. We show how it can be applied to cancer, heart disease, and infection. Here is an example of senescent-like foci in infarction spatial data (4/5)
24.02.2025 19:31 — 👍 0 🔁 0 💬 1 📌 0
These signatures recapitulate in vivo cellular senescence better than available gene sets derived from in vitro studies (3/5)
24.02.2025 19:31 — 👍 0 🔁 0 💬 1 📌 0We derive cell-type-specific weighted signatures of cellular senescence for humans and mice and universal signatures of genes enriched in multiple signatures. We combine these signatures with a scoring tool to identify senescence in your data. (2/5) github.com/jaleesr/SenePy
24.02.2025 19:31 — 👍 0 🔁 0 💬 1 📌 0
Senescent cells contribute to disease and are found in many tissues but are hard to analyze because markers have been derived in culture and do not account for cell-type differences. Here, we define new signatures based on millions of single cells (1/5) www.nature.com/articles/s41...
24.02.2025 19:31 — 👍 23 🔁 6 💬 1 📌 0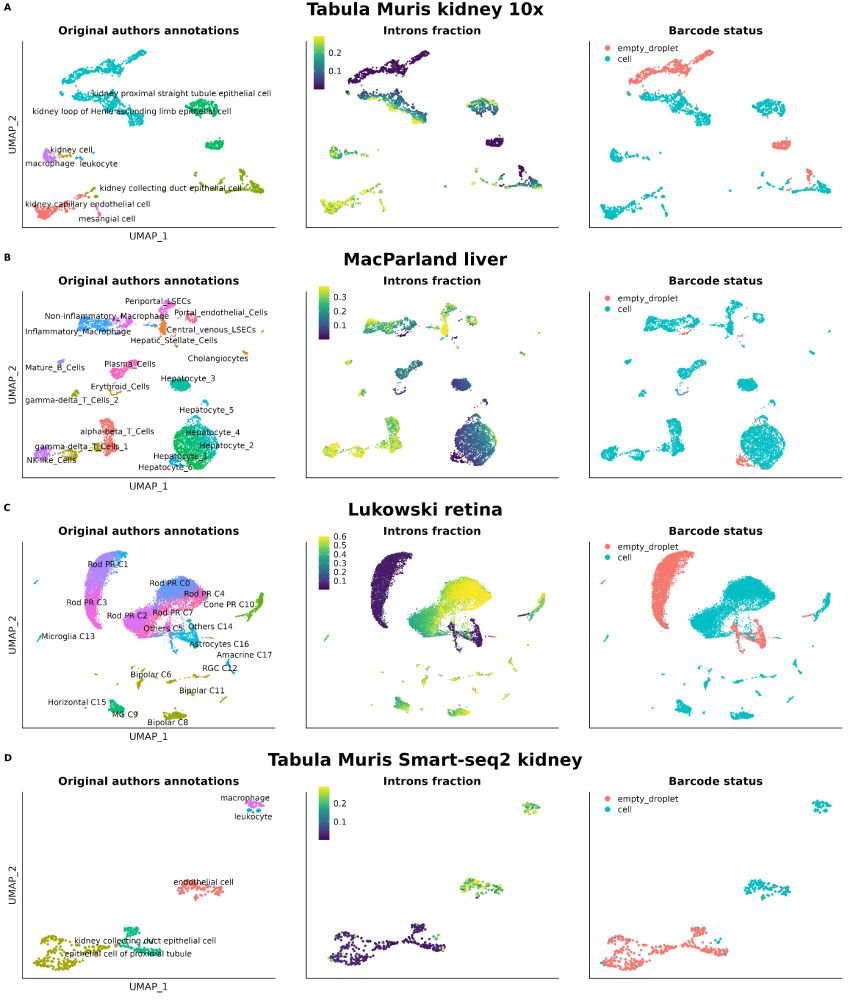
Not checking nuclear markers like MALAT1 or intronic reads in your scRNA-seq data?🚨
We show their power to flag low-quality cells—even in top public datasets. It’s time to prioritize better QC for cleaner, more reliable genomics research!
Read more: bmcgenomics.biomedcentral.com/articles/10....
1/8
Exactly. Also related is how most preprocessing workflows don’t account for cell types/condition and treat everything as one distribution.
24.11.2024 18:07 — 👍 1 🔁 0 💬 0 📌 0Should we instead normalize cell a/b to some shared technical/depth factor? Eg, cell A is 0.4x depth compared to other A cells. So scale cell B to 0.4x to other B cells. Then combine? In my mind this is more true to reality
24.11.2024 16:44 — 👍 0 🔁 0 💬 1 📌 0Doublet detection methods simulate doublets by adding or averaging exp in random cell pairs. But, there is technical variation between cells. A true doublet is 2+ cells processed in the same droplet with no technical variation. Eg, cell A (9000 UMI)+ cell B (1000 UMI) still looks like cell A.
24.11.2024 16:32 — 👍 1 🔁 0 💬 1 📌 0
Typical single-cell preprocessing can be unfair to certain cell types. For example, fewer genes are typically detected in neutrophils and they are often mistakenly removed.
I’ve made a short video covering this simple but important concept:
youtu.be/r4A_QgseUfw?...
There are so many people moving over that I'm sure I'm missing folks. Can we make a #compbio / #genomics intro thread to get reacquainted?
I'm at the University of Colorado. I often say that if you pick two of three from #transcriptome, #ML, and #publicdata, my lab is probably interested.

Mark(@sanbomics.bsky.social) put together a really nice video & walkthrough on using alevin-fry/simpleAF to process your single-cell RNA-seq data. If you're doing processing of such data, I recommend checking it out as a transparent & open alternative to CellRanger (& way faster) t.co/Hu9ueRduQ9! 🖥️🧬
15.09.2023 13:16 — 👍 20 🔁 11 💬 2 📌 0