Minimally this is a straightforward data pre-processing step that would probably improve the performance of all methods.As it is, I think the results are misleading because you suggest that bmws and other methods cannot estimate large selection coefficients which is not true.
07.02.2026 21:10 —
👍 0
🔁 0
💬 1
📌 0

I agree you shouldn't condition but I think if you see a trajectory like this you should remove the region in red from the inference. As you say, it's uninformative – the selection coefficient could be anything – and you do exclude the equally uninformative blue region.
07.02.2026 21:10 —
👍 0
🔁 0
💬 1
📌 0
Are the selected variants fixing in the longer time series? I think if you include long stretches where the allele frequency is 1 then bmws will underestimate the selection coefficient due to the shrinkage on s. But I think in practice you would exclude those observations.
04.02.2026 16:03 —
👍 1
🔁 0
💬 1
📌 0
Converse is people using the term "experiment" to describe some kind of data analysis.
23.10.2025 12:47 —
👍 1
🔁 0
💬 0
📌 0
And Mary-Claire
18.10.2025 21:33 —
👍 1
🔁 0
💬 1
📌 0
Well I suppose specifically for a developmental trait like this I might think that the environmental assumptions of the twin study design would be violated - i.e. twins share more of the relevant (in utero) environment than sibs, leading to an overestimate of h2.
18.10.2025 13:57 —
👍 1
🔁 0
💬 1
📌 0
I don't think this really makes sense though; it's like how anthropologists sometimes talk about things like "potential height" which is the height you would reach if you had perfect nutrition. But it's not a phenotype.
17.10.2025 22:38 —
👍 1
🔁 0
💬 1
📌 0
Indeed, rates of right-handedness were increased by [an environment] in which people were forced to write right-handed.
17.10.2025 13:27 —
👍 2
🔁 0
💬 1
📌 0
I mean if someone is forced to the point where they use their right hand all the time, are they not right-handed?
17.10.2025 13:25 —
👍 0
🔁 0
💬 1
📌 0
Yeah, those are always inflated
17.10.2025 13:24 —
👍 0
🔁 0
💬 1
📌 0
Also I don't believe the heritability is 25%. UKB SNP heritability is less than 2%.
17.10.2025 01:22 —
👍 0
🔁 0
💬 1
📌 0
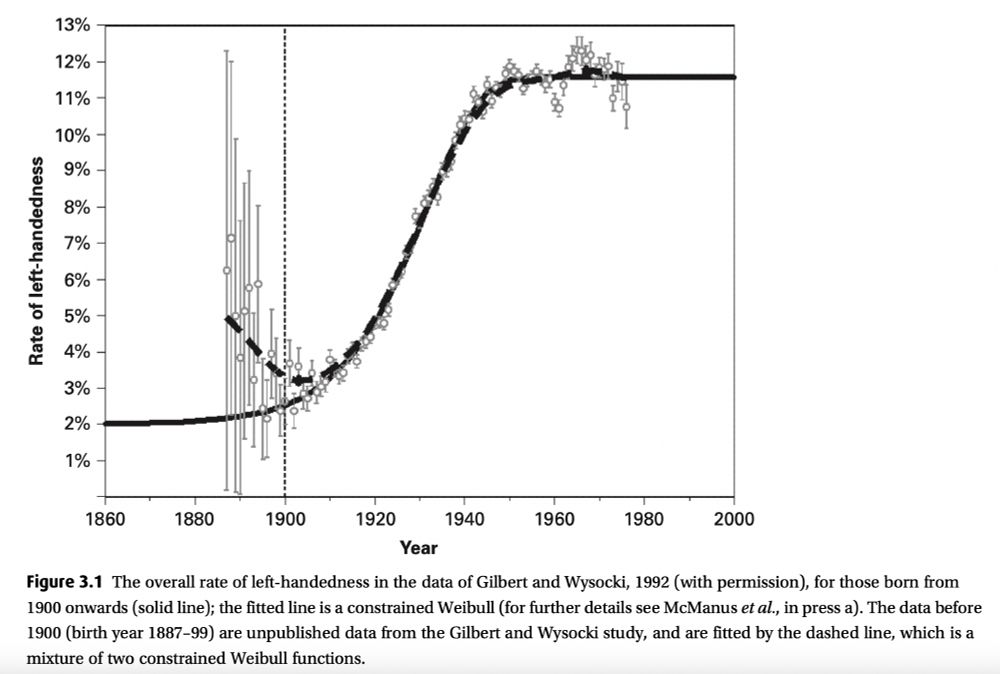
It's still environment, even if it's not practically measurable! Also, handedness can modified [by changing the environment].
17.10.2025 01:19 —
👍 4
🔁 0
💬 3
📌 0
Not sure the individual variation is the issue; it looks like they see it in most samples but I wonder whether there could be an off-target effect in the PCR step. Would want to check whether the putative mutant reads are enriched for mismatches.
12.09.2025 14:52 —
👍 1
🔁 0
💬 0
📌 0
The only bit of the ARG you own is your terminal branch.
02.08.2025 03:00 —
👍 3
🔁 0
💬 1
📌 0
We'll have to fix that!
31.07.2025 12:59 —
👍 1
🔁 0
💬 0
📌 0
I don't know if it's better. They can certainly phase better, and this suggests they have access to UKB data: www.ukbiobank.ac.uk/projects/hig.... Plus they have in-house non-EUR panels (e.g. www.nature.com/articles/s42...)
30.07.2025 02:36 —
👍 1
🔁 0
💬 1
📌 0
Yes seems so, at least a couple of months ago I was able to download imputed data.
29.07.2025 21:53 —
👍 1
🔁 0
💬 2
📌 0
True, I mean I guess that [people imputing their own genomes] is exactly what they don't want people doing. Also 23andMe gives you imputed data, possibly better than TopMed imputation anyway?
29.07.2025 12:56 —
👍 1
🔁 0
💬 1
📌 0
Maybe because they don't want people trying to split up their samples over many small jobs. If you just have one, I guess just add in 19 samples from 1000 Genomes?
29.07.2025 01:21 —
👍 2
🔁 0
💬 1
📌 0
He chose.... poorly
24.07.2025 15:41 —
👍 2
🔁 0
💬 1
📌 0
I find Figure 7 more informative about the route than Figure 1
24.07.2025 14:10 —
👍 1
🔁 0
💬 1
📌 0
I think it makes very little difference - the high LD regions are small (and don't obviously have different patterns of cross-individual differences), so won't have much effect on the genome-wide differences.
23.07.2025 16:33 —
👍 1
🔁 0
💬 0
📌 0
In practice the main use of LD pruning seems to be removing high ("long-range") LD regions that otherwise show up as individual PCs (e.g. see Fig1 etc here: academic.oup.com/bioinformati...). In some sense those are still "real" structure in the ARG, but it's kind of undesirable for some analyses.
22.07.2025 17:50 —
👍 5
🔁 1
💬 1
📌 0
I think you could probably bias it one way or another depending on how you do the LD pruning (LD in which population?). I think of it as like you're changing the relative weighting of different parts of the ARG.
22.07.2025 17:50 —
👍 0
🔁 0
💬 2
📌 0
So |EUR-YRI| can be < |YRI-X| for some other African population X, due to structure within Africa. |EUR-YRI| can be < |YRI-YRI| for some PCs but if you sum the distances across enough PCs, weighting them appropriately, it will always be larger.
21.07.2025 03:37 —
👍 1
🔁 0
💬 0
📌 0
I see, but to the original point that's not how PCA works. I don't think |EUR-YRI| is ever going to be less than |YRI-YRI| in PC space – again YRI-YRI can coalesce before the split. Of course if you only look at the first 2 (or first N) PCs, it can be, but that just depends on sampling.
21.07.2025 03:37 —
👍 2
🔁 0
💬 1
📌 0
They are not measuring pairwise differences. If I understand their figure 4A, if one individual is 0/0 and another is 1/0 then they count that as S=1. But If the second individual is 1/1 then they also count that as S=1 (not S=2). So they undercount differences in individuals with low heterozygosity
21.07.2025 01:59 —
👍 0
🔁 0
💬 1
📌 0
Another way to think about it: the variants that fix in A during the bottleneck are equally likely to fix in the matching or non-matching state as a randomly chosen individual from B, so the expected number of differences does not change.
20.07.2025 20:37 —
👍 4
🔁 0
💬 1
📌 0
On the other hand, Fst does behave the way you describe, because Fst between A and B depends on E[T_aa] – which does depend on what happens on the A lineage – as well as E[T_ab]. So Fst between A and B is > Fst between B and C.
20.07.2025 20:33 —
👍 2
🔁 0
💬 1
📌 0

