legend 2025 will be next Tuesday/Wednesday/Thursday.
The detailed program is on legend2025.sciencesconf.org?lang=en
If you are not attending the conference you can still follow the presentations online (Zoom links are on the website).
05.12.2025 11:45 — 👍 2 🔁 3 💬 0 📌 0
Very happy about this work on phylogenetic neural inference, led by @lblassel.bsky.social :)
17.10.2025 05:27 — 👍 7 🔁 2 💬 0 📌 0
![Exploring the space of self-reproducing RNA using generative models, Martin Weigt
Exploring the archaic introgression landscape of admixed populations through
joint ancestry inference, Jazeps Medina Tretmanis [et al.]
Predicting natural variation in the yeast phenotypic landscape with machine
learning, Sakshi Khaiwal [et al.]
Phylodynamic modeling with unsupervised Bayesian neural networks, Marino
Gabriele [et al.]
Likelihood-free inference of phylogenetic tree posterior distributions, Luc Blas-
sel [et al.]
Generative continuous time model reveals epistatic signatures in protein evolu-
tion, Barrat-Charlaix Pierre
Neural posterior estimation for high-dimensional genomic data from complex pop-
ulation genetic models, Jiseon Min [et al.]
A differentiable model for detecting diversifying selection directly from alignments
in large-scale bacterial datasets, Leonie Lorenz [et al.]
Detecting interspecific positive selection using transformers, Charlotte West [et al.]
Predicting Multiple Sequence Alignment Uncertainty via Machine Learning, Lucia
Martin-Fernandez [et al.]
Graph Neural Networks for Likelihood-Free Inference in Diversification Mod-
els, Amélie Leroy [et al.]
Popformer: learning general signatures of genetic variation and natural selection
with a self-supervised transformer, Leon Zong [et al.]](https://cdn.bsky.app/img/feed_thumbnail/plain/did:plc:7x5c6ifrcznmg4mzhfuc2a6r/bafkreidur7bz43o2jhwr4gbt36awozawqthbkkfeszxjkxf4kvhyh7q6du@jpeg)
Exploring the space of self-reproducing RNA using generative models, Martin Weigt
Exploring the archaic introgression landscape of admixed populations through
joint ancestry inference, Jazeps Medina Tretmanis [et al.]
Predicting natural variation in the yeast phenotypic landscape with machine
learning, Sakshi Khaiwal [et al.]
Phylodynamic modeling with unsupervised Bayesian neural networks, Marino
Gabriele [et al.]
Likelihood-free inference of phylogenetic tree posterior distributions, Luc Blas-
sel [et al.]
Generative continuous time model reveals epistatic signatures in protein evolu-
tion, Barrat-Charlaix Pierre
Neural posterior estimation for high-dimensional genomic data from complex pop-
ulation genetic models, Jiseon Min [et al.]
A differentiable model for detecting diversifying selection directly from alignments
in large-scale bacterial datasets, Leonie Lorenz [et al.]
Detecting interspecific positive selection using transformers, Charlotte West [et al.]
Predicting Multiple Sequence Alignment Uncertainty via Machine Learning, Lucia
Martin-Fernandez [et al.]
Graph Neural Networks for Likelihood-Free Inference in Diversification Mod-
els, Amélie Leroy [et al.]
Popformer: learning general signatures of genetic variation and natural selection
with a self-supervised transformer, Leon Zong [et al.]
![PRIVET: PRIVacy metric based on Extreme value Theory, Antoine Szatkownik [et
al.]
Generative models for inferring the evolutionary history of the malaria vector
Anopheles gambiae, Amelia Eneli [et al.]
Language Models Outperform Supervised-Only Approaches for Conserved Ele-
ment Comprehension, Eyes Robson [et al.]
Identification and Classification of Orphan Genes, Spurious Orphan Genes, and
Conserved Genes from the human microbiome, Chen Chen
Neural Simulation-based inference of demography and selection, Francisco De
Borja Campuzano Jiménez [et al.]
Species Identification and aDNA Read Mapping Using k-mer Embeddings, Filip
Thor [et al.]
Contrastive Learning for Population Structure and Trait Prediction, Filip Thor [et
al.]
Protein and genomic language models chart a vast landscape of antiphage de-
fenses, Mordret Ernest
The Phylogenomics and Sparse Learning of Trait Innovations, Gaurav Diwan [et
al.]](https://cdn.bsky.app/img/feed_thumbnail/plain/did:plc:7x5c6ifrcznmg4mzhfuc2a6r/bafkreig4akadduemzbit67bv7jnr7x2ircwiriykabg2imuf3phy6hhube@jpeg)
PRIVET: PRIVacy metric based on Extreme value Theory, Antoine Szatkownik [et
al.]
Generative models for inferring the evolutionary history of the malaria vector
Anopheles gambiae, Amelia Eneli [et al.]
Language Models Outperform Supervised-Only Approaches for Conserved Ele-
ment Comprehension, Eyes Robson [et al.]
Identification and Classification of Orphan Genes, Spurious Orphan Genes, and
Conserved Genes from the human microbiome, Chen Chen
Neural Simulation-based inference of demography and selection, Francisco De
Borja Campuzano Jiménez [et al.]
Species Identification and aDNA Read Mapping Using k-mer Embeddings, Filip
Thor [et al.]
Contrastive Learning for Population Structure and Trait Prediction, Filip Thor [et
al.]
Protein and genomic language models chart a vast landscape of antiphage de-
fenses, Mordret Ernest
The Phylogenomics and Sparse Learning of Trait Innovations, Gaurav Diwan [et
al.]
The decisions for LEGEND are out: legend2025.sciencesconf.org/data/book_le...
I'm really looking forward to hearing these 21 exciting presentations (and additional 30 posters) next December.
If you want to attend too, registration is open until October 17th through legend2025.sciencesconf.org
08.10.2025 11:04 — 👍 4 🔁 1 💬 1 📌 0

Only a few hours left to submit your abstract for a talk at the Machine Learning in Evolutionary Genomics conference in December in Aussois in the French alps!
22.09.2025 14:46 — 👍 2 🔁 2 💬 0 📌 0

MLCB - Schedule
The in-person component will be held at the New York Genome Center, 101 6th Ave, New York, NY 10013.
#MLCB2025 is tomorrow & Thursday with a fantastic lineup of keynotes & contributed talks www.mlcb.org/schedule. We'll be livestreaming through our YouTube channel www.youtube.com/@mlcbconf. Thanks to www.corteva.com, instadeep.com, the Simons Center at CSHL & NYGC for generous support!
10.09.2025 00:16 — 👍 9 🔁 2 💬 0 📌 0


Achievement unlocked: defend your habilitation thesis on the same day than your partner. That was quite a science + celebration day, thanks to all involved 💙✨
05.09.2025 16:51 — 👍 45 🔁 7 💬 6 📌 0

🌎👩🔬 For 15+ years biology has accumulated petabytes (million gigabytes) of🧬DNA sequencing data🧬 from the far reaches of our planet.🦠🍄🌵
Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open.
doi.org/10.1101/2024...
03.09.2025 08:39 — 👍 218 🔁 118 💬 3 📌 16
The call for abstract for LEGEND is now open:
legend2025.sciencesconf.org
It will close on September 22nd (oral presentations) and October 1st (posters).
Send us your best work on Machine Learning for Evolutionary Genomics and come discuss it with us in the French Alps next December!
02.09.2025 06:51 — 👍 3 🔁 3 💬 0 📌 0

#TalentCNRS 🥉| Flora Jay, entre génomes synthétiques et récits évolutifs, reçoit la médaille de bronze du CNRS.
➡️ www.ins2i.cnrs.fr/fr/cnrsinfo/...
🤝 @lisnlab.bsky.social @cnrs-paris-saclay.bsky.social
21.07.2025 12:01 — 👍 5 🔁 4 💬 0 📌 0
Preprint alert! 🦌
Our new abundance index, REINDEER2, is out!
It's cheap to build and update, offers tunable abundance precision at kmer level, and delivers very high query throughput.
Short thread!
www.biorxiv.org/content/10.1...
github.com/Yohan-Hernan...
19.06.2025 09:12 — 👍 23 🔁 13 💬 1 📌 4
Registration is now open!
The 580€ include housing and all meals.
We will close on October 17th or when reaching 80 participants.
18.06.2025 07:22 — 👍 4 🔁 4 💬 0 📌 0
Home - ProbGen 2026
Your Site Description
The 2026 Probabilistic Modeling in Genomics (ProbGen) meeting will be held at UC Berkeley, March 25-28, 2026. We have an amazing list of keynote speakers and session chairs:
probgen2026.github.io
Please help spread the news.
06.06.2025 17:52 — 👍 69 🔁 36 💬 2 📌 0
Merci à @cnrs-rhoneauvergne.bsky.social et @astropierre.com pour cette interview sur mes travaux en IA pour la génomique évolutive!
02.06.2025 10:53 — 👍 15 🔁 3 💬 0 📌 1
There is a nice example in @stephaneguindon.bsky.social's Ph.D thesis p.55
theses.hal.science/tel-00843343...
03.04.2025 12:29 — 👍 0 🔁 0 💬 0 📌 0
The design matrix of the regression should be nPairs x nBranches, and have a 1 at coordinates (i,j) such that branch j belongs to the path defined by pair i in the tree, 0 otherwise.
03.04.2025 12:26 — 👍 0 🔁 0 💬 1 📌 0
I think one way to do this is the least squares method, which gives you the set of branch lengths on your given topology such that the sum of squared differences between your given distances and the distances on the tree are minimal.
03.04.2025 12:23 — 👍 0 🔁 0 💬 1 📌 0
Phyloformer is finally published in MBE! 🎉
academic.oup.com/mbe/advance-...
The thread below provides a summary of our neural network for likelihood-free phylogenetic reconstruction.
12.03.2025 11:49 — 👍 16 🔁 11 💬 0 📌 1

People having breakfast in front of the Alps in the Centre Paul Langevin.
Come hear about the latest advances in the field and discuss your own work at Centre Paul Langevin in beautiful Aussois.
24.02.2025 08:58 — 👍 1 🔁 0 💬 0 📌 0

A headshot of Dr Burak Yelmen.
Burak Yelmen from the University of Tartu will give a keynote presentation on "A perspective on generative neural networks in genomics with applications in synthetic data generation".
24.02.2025 08:58 — 👍 1 🔁 1 💬 1 📌 0

A headshot of Dr Claudia Solis-Lemus.
Claudia Solís-Lemus from the University of Wisconsin-Madison will give a keynote presentation on "The good, the bad and the ugly of deep learning in phylogenetic inference".
24.02.2025 08:58 — 👍 2 🔁 1 💬 1 📌 0

A headshot of Dr Anne-Florence Bitbol.
Anne-Florence Bitbol from EPFL will give a keynote presentation on "Coevolution-aware language models".
24.02.2025 08:58 — 👍 2 🔁 1 💬 1 📌 0

A legendary being holds a phylogenetic tree in the palm of their hand, with snowy mountains in the background.
The next LEGEND conference on machine learning for evolutionary genomics will be in Aussois (French Alps) between December 8th and 12th.
Mark your calendars and make sure your best work is ready next September when the call for abstracts opens 🙂
legend2025.sciencesconf.org
24.02.2025 08:58 — 👍 10 🔁 7 💬 1 📌 3

My book is (at last) out, just in time for Christmas!
A blog post to celebrate and present it: francisbach.com/my-book-is-o...
21.12.2024 15:23 — 👍 142 🔁 35 💬 2 📌 3
Ok, I tried to create my own list of people working on developing statistical or machine learning models applied to omics data. I am sure I missed a lot of cool people. If you'd like to be added, let me know. #Stats #ML #Omics
go.bsky.app/73rcuJn
24.11.2024 07:50 — 👍 95 🔁 36 💬 38 📌 4
Hi Raphael, thanks for putting this together, I'll be happy to be in the list if you think it makes sense :)
26.11.2024 08:05 — 👍 0 🔁 0 💬 0 📌 0
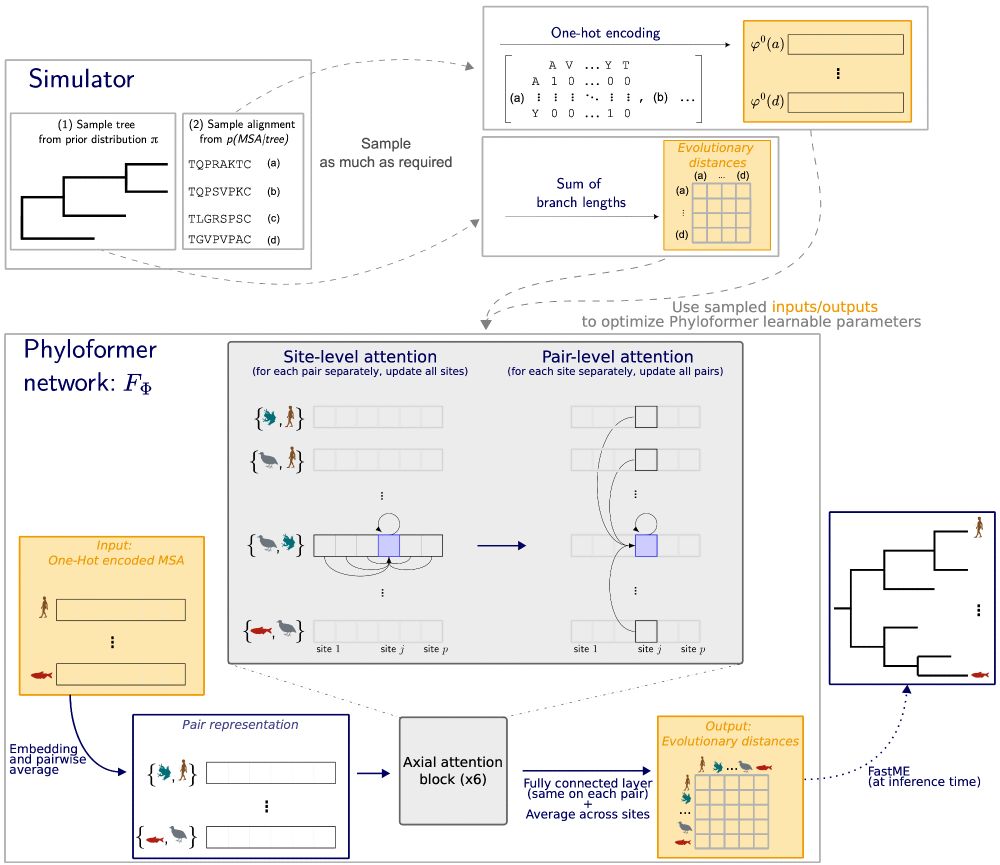
A sketch summarizing the entire Phyloformer process.
All this work was done by Luca Nesterenko and
@lblassel.bsky.social , assisted by P. Veber, Bastien Boussau
and myself.
The code and data are available at github.com/lucanest/Phy...
Please share if you find this interesting, and we welcome your feedback :)
24.06.2024 08:35 — 👍 1 🔁 1 💬 0 📌 0
machine learning researcher @ Apple machine learning research
Genetics, bioinformatics, comp bio, statistics, data science, open source, open science!
Evolutionary genomics, nearly-neutralist...
Structural Phylogenetics • methods, datasets, tools, events • concise updates for researchers and practitioners. Managed by @proteinmechanic.bsky.social and @cpuentelelievre.bsky.social
P.A. Chaize IRL
Historien "martial studies" (pour les académiques)
Records Manager avec accent
Mec de droite qui dit des trucs de gauche.
Echelle de confiance :
AMHE: 9/10
Histoire médiévale et moderne: 5 à 8/10
W40k: 4/10
Le reste ? MHO
Compte officiel du Laboratoire de Biométrie et Biologie Evolutive, Lyon, France.
Official account of the Laboratory of Biometry and Evolutionary Biology, Lyon, France.
Professor detached@CNRS/Sorbonne Université, ERC and IUF fellow, co-founder@qubit_pharma, structural bioinformatics, Chemoinformatics, molecular visualization
The 2026 edition will be held in Lille (France) on 7-9 January ! https://populationgeneticsgroup.org.uk/
Professor at U. Regensburg, Germany #Ecology #Evolution #Statistics #MachineLearning
https://www.uni-regensburg.de/biologie-vorklinische-medizin/forschen/arbeitsgruppen/ag-hartig
PhD student at Université de Montpellier. Community ecologist.
Économiste (Ass. prof.) - Université Paris 13 - CEPN - Santé, sécurité sociale, capitalisme
https://cepn.univ-paris13.fr/nicolas-da-silva/
https://lafabrique.fr/la-bataille-de-la-secu/
https://www.alternatives-economiques.fr/users/nicolas-da-silva
In Slumberland, obviously
Bioinformatics Scientist @pasteur.fr , #virus, #sequencing, #phylogeny, #bioinformatics
Collectif de lutte contre les violences sexistes et sexuelles dans l’enseignement supérieur.
⚡️ clasches.fr | clasches@proton.me
Virus evolution | genomic epidemiology| virus origins | phylogenetics | BEAST | ARTIC Network
University of Edinburgh
http://beast.community
http://artic.network
VIB Center for AI & Computational Biology
🧑💻 Director @steinaerts.bsky.social
📣 We're recruiting group leaders vib.ai
📻 Follow us for all the latest updates
Computational biologist interested in deciphering the genomic regulatory code at vib.ai
The world's leading publication for data science and artificial intelligence professionals.
Website 🌐 towardsdatascience.com
Submit an Article ✍️ https://contributor.insightmediagroup.io
Subscribe to our Newsletter 📩 https://bit.ly/TDS-Newsletter