Are you interested in doing a PhD in Copenhagen? Interested in studying Neanderthals and Denisovans which live on in our genomes?
Than you are more than welcome to apply to join my group starting Jan 2026 :)
candidate.hr-manager.net/ApplicationI...
Please reach out if you have any questions!
28.08.2025 12:35 —
👍 58
🔁 44
💬 1
📌 5

Happy to share a PhD opportunity in my new lab at the University of Zurich! Application deadline: June 30. Do not hesitate to contact me for any questions!
20.05.2025 12:19 —
👍 19
🔁 25
💬 1
📌 0
And thanks to Lidia Gonfaus for making this beautiful illustration! :)
05.05.2025 14:26 —
👍 2
🔁 0
💬 0
📌 0
We hope this method will be useful to people and look forward to hearing your feedback!
05.05.2025 13:34 —
👍 1
🔁 0
💬 0
📌 0

We also found cases where hybrid decoding recovers a putative neanderthal fragment that was split in two by posterior decoding and where only half was found by viterbi decoding.
05.05.2025 13:34 —
👍 2
🔁 0
💬 1
📌 0
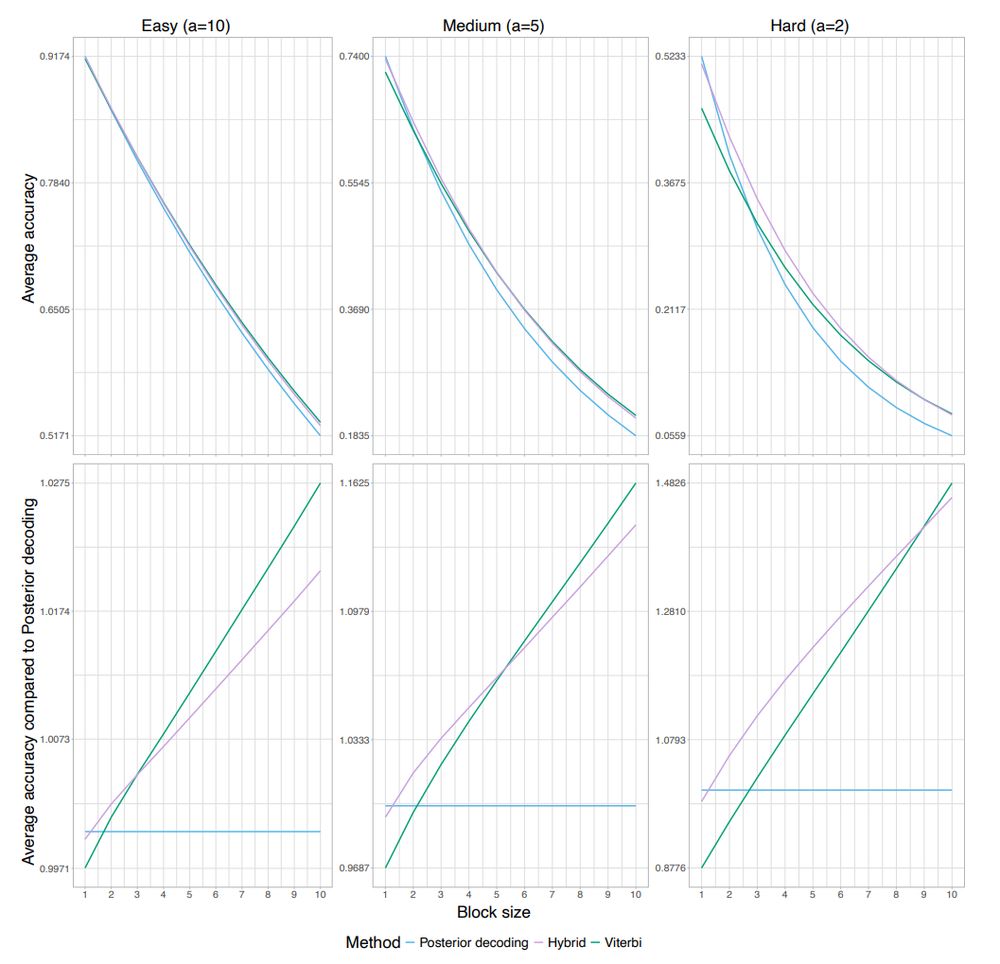
In practice this hybrid decoding has the highest accuracy when it comes to intermediate sized fragments. For short fragment posterior decoding is best and for long fragments viterbi is best.
05.05.2025 13:34 —
👍 0
🔁 0
💬 1
📌 0

We recommend picking the alpha closest to the 45 degree line (ideally you want to be as close to the top right corner as possible). We call these plots Artemis plots because of the bow like shape!
05.05.2025 13:34 —
👍 0
🔁 0
💬 1
📌 0
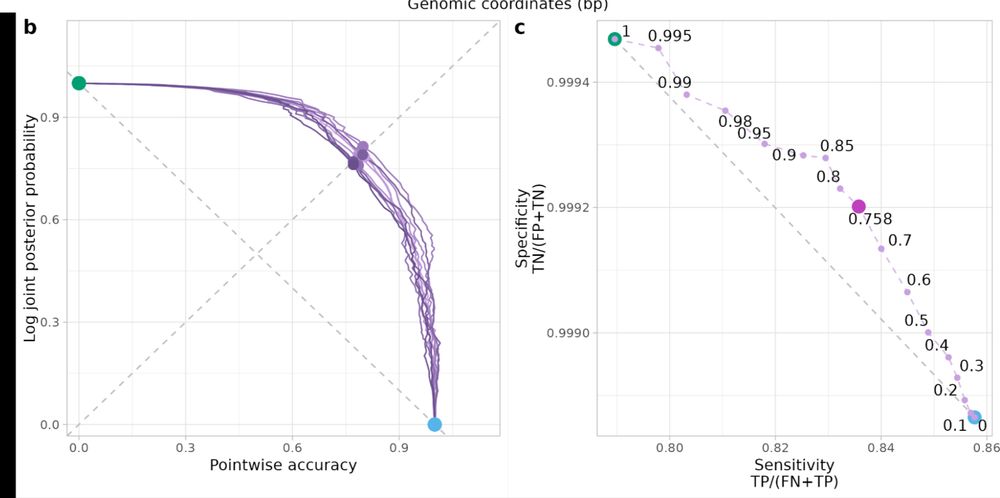
If you simulate data from your emission and transition parameters you can calculate point wise accuracy/joint posterior probability and sensitivity/specificity plots for each alpha and pick the best one!
05.05.2025 13:34 —
👍 0
🔁 0
💬 1
📌 0
The best alpha will depend on the specific emission and transition parameters. Also an alpha of 0.5 does not nescessaraly mark the halfway point between the two methods. There we propose a simulation approach for picking the appropriate alpha for your analyse.
05.05.2025 13:34 —
👍 0
🔁 0
💬 1
📌 0

If only there was a way to have your cake and eat it too! Inspired by previous work we propose a weighted mean of posterior and viterbi decoding controlled by a parameter alpha. When alpha = 0 you get posterior decoding and when alpha = 1 you get viterbi. When alpha is intermediate you get a mix.
05.05.2025 13:34 —
👍 0
🔁 0
💬 1
📌 0
Posterior decoding maximixes the local probability - that is the pointwise accuracy. This leads to more short fragments being recovered but also a higher false positive rate.
05.05.2025 13:34 —
👍 0
🔁 0
💬 1
📌 0
Viterbi decoding maximixes the global probability of the hidden state path. While leading to high accuracy this decoding tends to not switch states as often as it should and many short segments will be lost.
05.05.2025 13:34 —
👍 0
🔁 0
💬 1
📌 0
This part of the manuscript has to do with finding the best decoded path for a hidden markov model so let's talk the two most used: Viterbi and posterior decoding
05.05.2025 13:34 —
👍 0
🔁 0
💬 1
📌 0
We ( @zeniabaek.bsky.social @moicoll.bsky.social and @asgerhobolth.bsky.social ) present a new cool way to visualize the optimal trade off for hmm decoding called Artemis plots!
arxiv.org/pdf/2504.15156
www.biorxiv.org/content/10.1...
05.05.2025 13:34 —
👍 20
🔁 9
💬 2
📌 0
If you haven't already you can go follow the amazing:
@moicoll.bsky.social , @zeniabaek.bsky.social and @asgerhobolth.bsky.social :)
02.05.2025 14:20 —
👍 0
🔁 0
💬 0
📌 0
The analytical is based on finite markov chain embedding :) The bimodal longest has to do with fact that in sometimes the longest fragments breaks in two!
01.05.2025 21:39 —
👍 1
🔁 0
💬 0
📌 0
This was a great collaboration with Moisès Coll Macià, Zenia Elise Damgaard Bæk and Asger Hobolth :)
01.05.2025 13:12 —
👍 2
🔁 0
💬 1
📌 0
While this approach is useful for obtaining summary statistics it is not good at finding a single good hidden state path through the genome like Viterbi/posterior decoding. We have some thought on that too but that is for another thread!
01.05.2025 13:12 —
👍 2
🔁 0
💬 1
📌 0
This sampling approach is easy to implement and can be used for all HMMs commenly used! One could calculate what the distribution of long IBD fragments are between two ancient individuals, how much time you are in a given TMRCA state for PSMC or sample paths through the Li
-Stephens copying model.
01.05.2025 13:12 —
👍 4
🔁 0
💬 1
📌 0

We also show that our estimates of Neanderthal ancestry is consistent with f4 ratio estimates and we recover the signal of different archaic fragment length distributions across continental groups! We explored the explanations for this signal previously (www.nature.com/articles/s41...)
01.05.2025 13:12 —
👍 2
🔁 0
💬 1
📌 0

You can even get close to the true fragment length distribution. Black vertical line is true mean fragment length and black curve is expected theoretical distribution. Colored dotted vertical lines are means for each decoding type
01.05.2025 13:12 —
👍 2
🔁 0
💬 1
📌 0

We show that by sampling hidden state sequences you can accurately recover summmary statistics (black dotted line) such as: How much of our genome is archaic? How many archaic fragments of DNA is there? How long is the longest fragment? You can even do it analytically! (red curve)
01.05.2025 13:12 —
👍 3
🔁 0
💬 2
📌 0

Another strategy is to sample many decoded paths conditioned on the data. The benefit of this is that sometimes you find the short fragments! And instead of one decoded path you have many sampled paths from which you can calculate any summary statistic you want (with confidence intervals!)
01.05.2025 13:12 —
👍 2
🔁 0
💬 1
📌 0
Decoding is not perfect and we cannot find all the archaic DNA fragments our genomes - especially when they are short. For decoding people typically use Viterbi decoding (more accurate but misses many archaic fragments) or Posterior decoding (less accurate but finds more archaic fragments)
01.05.2025 13:12 —
👍 2
🔁 0
💬 1
📌 0

To find fragments we use is a Hidden Markov Models (HMMs) which classifies the genome into an archaic and a human state based on the SNP-density of variants not found in African genomes where there is little archaic DNA. We then "decode" the genome to find into archaic and human fragments.
01.05.2025 13:12 —
👍 1
🔁 0
💬 2
📌 0
Archaic DNA exists in small fragments throughout your genome if we are good at finding all of them we could answers questions like:
How much of our genome did we get from Neanderthals/Denisovans? When did we meet them (shorter fragments of DNA = long ago, long fragments of archaic DNA = recent)?
01.05.2025 13:12 —
👍 3
🔁 0
💬 1
📌 0
Very excited to be part of two manuscripts which are now on biorxiv!
arxiv.org/pdf/2504.15156
www.biorxiv.org/content/10.1...
Archaic humans such as Neanderthals and Denisovans are extinct but their DNA lives on in many humans today because our ancestors met and had children with them.
01.05.2025 13:12 —
👍 28
🔁 10
💬 1
📌 0