
This wouldn’t have been possible without your support and guidance, grateful to have benefitted from that!
05.03.2026 15:16 — 👍 0 🔁 0 💬 0 📌 0This wouldn’t have been possible without your support and guidance, grateful to have benefitted from that!
05.03.2026 15:16 — 👍 0 🔁 0 💬 0 📌 0
New pre-print identifying 29 Covid GWAS variants that drive allele-specific activity in a functional genomics screen in lung cells, from @gweykopf.bsky.social and
@eliasfriman.bsky.social @simonbiddie.bsky.social
www.biorxiv.org/content/10.6...
@uoe-igc.bsky.social
@uk-fgx.bsky.social
A massive thank you to my fabulous supervisors @eliasfriman.bsky.social @simonbiddie.bsky.social @wbickmor.bsky.social 🎉
10/10
In summary, combining high-throughput allele-specific screening with deep learning models, we identify COVID-19 risk variants with regulatory function in lung epithelial cells and propose testable hypothesis for how these impact COVID-19-linked pathways. We hope to inspire further follow up 🔎
9/10


We propose rs2297480 increases expression of an alternative FDPS isoform lacking part of the catalytic domain of farnesyl pyrophosphate synthase that could impact protein prenylation, including that of Rab GTPases which control the endolysosomal pathways used by SARS-CoV-2 for cellular entry 🦠
8/10


For example, we propose rs6517156, located in the last intron of the type I interferon receptor IFNAR2, which is crucial for the immune response to viruses, causes loss of enhancer activity by disrupting a p53 binding site.
7/10
But can these models still provide additional information? Yes, they can! Predicting the effect of amVars on chromatin features and in-silico mutagenesis with motif matching can generate hypothesis about endogenous effects, candidate transcription factors and target genes 🛠️
6/10
Can state-of-the-art deep learning models predict our experimentally identified enhancer activity-modulating (amVars) variants? Using Malinois and AlphaGenome, neither model predicts enhancer modulating variants well ❌
5/10

Interestingly, we identify 5 variant pairs within STARR-seq active enhancers at the KANSL1 risk locus, highlighting many variants can be functional at one cis-regulatory element (CRE) and at different CREs across one locus 💡
4/10

What if multiple variants in combination 🧬🧬 have effects beyond those observed for each variant in isolation? We tested ~3.8K combinations of variants occurring in close genomic proximity, finding both additive and non-additive effects (greater than expected loss) on enhancer activity
3/10

Testing ~5,000 COVID-19-associated variants using STARR-seq in lung epithelial cells alone and in combination, we find 166 variants within STARR-seq active sequences, 29 of which change STARR-seq activity (i.e. loss/gain) ⬆️⬇️
2/10
*New pre-print* I’m delighted to share the second story from my PhD 🥳 We identify 29 severe COVID-19-associated human genetic risk variants that modulate enhancer activity in lung epithelial cells
1/10
www.biorxiv.org/cgi/content/...
Thank you so much Christa!
15.11.2025 08:08 — 👍 0 🔁 0 💬 0 📌 0

A personal milestone - PhD successfully defended (with no corrections 🥳)
A heartfelt thank you to my supervisors @wbickmor.bsky.social @simonbiddie.bsky.social @eliasfriman.bsky.social @KennyBaillie and the entire Bickmore lab who made this a wonderful 4 years ✨
Find more details in the pre-print:
https://www.biorxiv.org/content/10.1101/2025.07.24.666385v3.full.pdf
14/
Huge thank you to my wonderful supervisors, all contributors and our fantastic collaborators who made this work possible. @wbickmor.bsky.social @simonbiddie.bsky.social @eliasfriman.bsky.social, @mbadonyi.bsky.social @leemurphy.bsky.social, Joe Marsh, Mark Gorrell, Jasmine Nguyen and others
13/
Studying genetic risk variants? Make sure to check for alternative isoforms using long-read RNA-seq data!
Our findings highlight a largely unexplored mechanism where disease-associated genetic variants act by causing damaging mutations in tissue-specific alternative isoforms. 12/
To recap, the DPP9 variant, previously thought to be non-coding, results in a likely structurally-damaging missense mutation in a lung-specific, functional, alternative DPP9 isoform. 11/
30.07.2025 11:09 — 👍 0 🔁 0 💬 1 📌 0
What is the impact of the COVID-19 variant? It causes a leucine to proline missense mutation in an N-terminal extension formed by the alternative exon, whereby the variant is predicted to disrupt an alpha-helix. 10/
30.07.2025 11:09 — 👍 0 🔁 0 💬 1 📌 0What do we know about DPP9’s function? It is a serine protease with diverse functions, including regulating the immune response by binding to and inhibiting the NLRP1 inflammasome. So is the alternative DPP9 isoform functional? Yes! It retains both enzymatic activity and NLRP1 binding. 9/
30.07.2025 11:09 — 👍 1 🔁 0 💬 1 📌 0
Is this novel variant-harbouring DPP9 isoform translated? Yes! The isoform is expressed and highly lung-specific 8/
30.07.2025 11:09 — 👍 0 🔁 0 💬 1 📌 0
How can we be confident this isoform is expressed? We developed a protocol for target-enriched long-read RNA-sequencing to achieve high coverage of even rare transcripts. We find fl-DPP9-AFE is significantly expressed in multiple lung cell lines. 7/
30.07.2025 11:09 — 👍 0 🔁 0 💬 1 📌 0
How about examples? We intersected GWAS variants for severe COVID-19 with alternative exons to identify a common variant located in an alternative first exon of an unannotated DPP9 isoform (fl-DPP9-AFE). 6/
30.07.2025 11:09 — 👍 0 🔁 0 💬 1 📌 0How can we assess the impact of variants on these alternative isoforms? We use AlphaFold structural modelling and variant effect prediction to estimate changes to the folding energy (ΔΔG), focusing on missense variants. 5/
30.07.2025 11:09 — 👍 0 🔁 0 💬 1 📌 0
Why do we think these are important? We find many of these isoforms are highly tissue-specific, more so than the corresponding reference isoform. For example, alternative isoforms harbouring respiratory trait-associated variants are frequently expressed in lung. 4/
30.07.2025 11:09 — 👍 0 🔁 0 💬 1 📌 0
So how frequent are variants in alternative isoforms? More common than we thought! Variants are more frequent in alternative than reference exons, in agreement with reduced selection pressure. 3/
30.07.2025 11:09 — 👍 0 🔁 0 💬 1 📌 0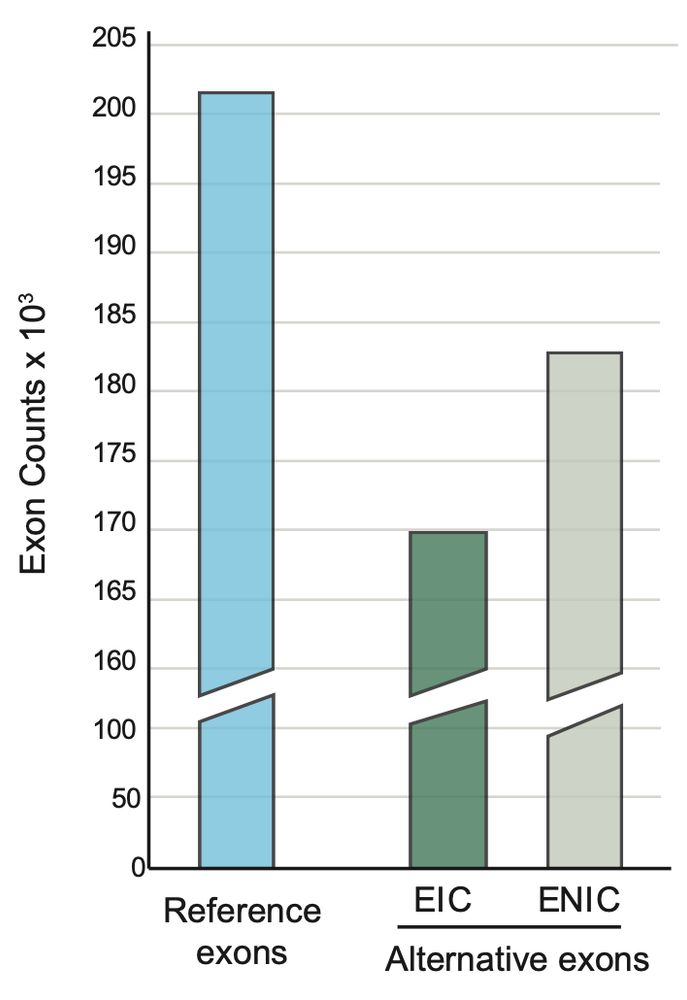
Genetic variants are typically interpreted by considering only reference isoforms. Yet the vast majority of human genes has multiple isoforms. In fact, based on long-read transcriptomic data, there is 350k alternative exons, ~half of which are not annotated by reference databases (ENIC).
30.07.2025 11:09 — 👍 0 🔁 0 💬 1 📌 0
Genetic variants can cause protein-coding mutations that result in disease. What about in alternative isoforms? Excited to share my first first-author pre-print! Here we show many variants reside in tissue-specific isoforms, causing coding mutations. 1/
https://bit.ly/455I6T6
Studying genetic risk variants? Make sure to check for alternative isoforms using long-read RNA-seq data!
Our findings highlight a largely unexplored mechanism where disease-associated genetic variants act by causing damaging mutations in tissue-specific alternative isoforms. 12/
To recap, the DPP9 variant, previously thought to be non-coding, results in a likely structurally-damaging missense mutation in a lung-specific, functional, alternative DPP9 isoform. 11/
30.07.2025 11:05 — 👍 0 🔁 0 💬 1 📌 0