

Exploring infection biology with mass spectrometry-based proteomics #MolOmics academic.oup.com/molecular-om...
19.02.2026 21:54 — 👍 2 🔁 1 💬 0 📌 0
Exploring infection biology with mass spectrometry-based proteomics #MolOmics academic.oup.com/molecular-om...
19.02.2026 21:54 — 👍 2 🔁 1 💬 0 📌 0
PTMOverlay: A Proteomic Tool to Visualize Post-Translational Modifications Across Evolution www.biorxiv.org/cont...
---
#proteomics #prot-preprint

LFQ Benchmark Dataset - Generation Beta: Assessing Modern Proteomics Instruments and Acquisition Workflows with High-Throughput LC Gradients www.biorxiv.org/cont...
---
#proteomics #prot-preprint

Wondering how cell surface proteins dynamically interact in response to signals? Check out this exciting study from Rasha Al Mismar and Anne-Claude Gingras that unveils new extracellular receptor partnerships and expands the toolkit for membrane interactomes: pubmed.ncbi.nlm.nih.gov/39499777/
30.01.2026 08:16 — 👍 1 🔁 1 💬 0 📌 0
- or even that we all know calculating confidence of peptide probabilities or gods forbid FDR is all over the place in different software we use daily
- do we know the math behind each one, or do we have a gold standard we benchmark them against (probably the cited paper did, so maybe)?
4/7

IsoPS-DIA: Dual Functionality of Absolute Targeted Quantification and Global Proteome Profiling pubs.acs.org/doi/10....
---
#proteomics #prot-paper

Trypsin is the workhorse of bottom-up #proteomics. But when was it first discovered? Lets try an emoji poll !
😇 1836
🤭 1876
😜 1897
🤑 1931
Reply with your guess!
Correct answer in the replies!
#TeamMassSpec
One of the things that I find odd about academics is that even though they often have only had one job that their supervisor arranged for them, they talk about employment related matters with a confidence that one might assume was due to vast experience.
20.09.2025 15:06 — 👍 3 🔁 1 💬 1 📌 0
DIA, DOA, DUI, DDA, etc. Here is a comparisons of some quantitative proteomics methods from a POV you might not have seen before:
github.com/pwilmart/qua...
Oh yes, the URL of the peptide mapper:
phbuffers.org/Claude/pepti...
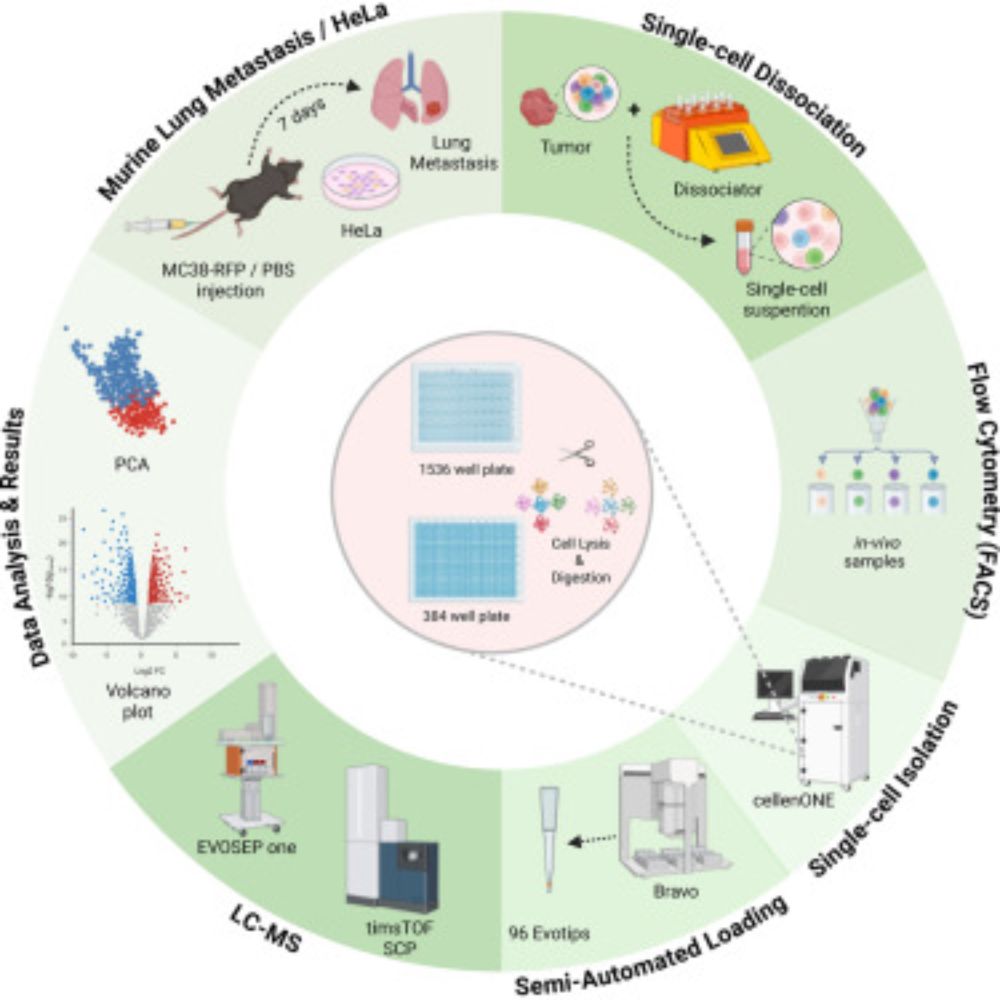
High throughput single-cell proteomics of in vivo cells #MCP #MassSpec www.mcponline.org/article/S153...
21.06.2025 07:00 — 👍 3 🔁 3 💬 0 📌 0
"More" IS "Better", no?
(\sarcasm)

Enhancing Sensitivity in Low-Load Proteomics Orbitrap Workflows via SLIM Integration #AC pubs.acs.org/doi/10.1021/...
11.06.2025 20:06 — 👍 1 🔁 1 💬 0 📌 0
Data Independent Acquisition to Inform the Development of Targeted Proteomics Assays Using a Triple Quadrupole Mass Spectrometer #JProteomeRes pubs.acs.org/doi/10.1021/...
07.05.2025 08:27 — 👍 3 🔁 3 💬 0 📌 0
Want to know how the ligands interact with proteins beyond model cell lines, e.g., in tissues or bacteria? Interested in membrane targets? Check out our High-Throughput PELSA method which allows you do all these cool screenings for dozens of ligands within two hours! www.biorxiv.org/content/10.1...
29.04.2025 18:13 — 👍 32 🔁 16 💬 1 📌 1🤣
09.04.2025 22:01 — 👍 0 🔁 0 💬 0 📌 0
(BioRxiv All) Development and Clinical Evaluation of a Multiplexed Health Surveillance Panel Using Ultra High-Throughput PRM-MS in an Inflammatory Bowel Disease Cohort.: Despite advances in clinical proteomics, translating protein biomarker discoveries into clinical use… #BioRxiv #MassSpecRSS
04.04.2025 18:14 — 👍 0 🔁 1 💬 0 📌 0Don't think it's relevant but "praja" in Sanskrit means "citizen" 🤣
01.04.2025 21:00 — 👍 2 🔁 1 💬 0 📌 0Missed writing "non-redundant" tryptic peptides
21.03.2025 23:43 — 👍 0 🔁 0 💬 1 📌 0Interesting. I wonder what this would look like if the cutoff scores were plotted against # tryptic peptides rather than # of entries in the FASTA.
21.03.2025 23:33 — 👍 0 🔁 0 💬 1 📌 0
Significant impact of consumable material and buffer composition for low-cell number proteomic sample preparation chemrxiv.org/engage/...
---
#proteomics #prot-preprint

Awesome paper, where we present and evaluate the methodology that will someday enable real-time monitoring of circulating biomarkers. pubs.acs.org/doi/full/10....
21.03.2025 03:20 — 👍 5 🔁 4 💬 0 📌 0
In-Depth Comparison of Reagent-Based Digestion Methods and Two Commercially Available Kits for Bottom-Up Proteomics pubs.acs.org/doi/ful...
---
#proteomics #prot-paper
It feels a little bit postclimactic to post this now, after the first version of our paper hit bioRxiv when X was still a thing that people used, the revised version was formally accepted at Cell Community past November. But here is the final print version of our paper
www.cell.com/iscience/ful...

Have a protein of unknown function? Try this www.tatta.bio/blog/gaia ! This is imo one of the best ai tools I have seen since alphafold alpha fold. I recommend you look at the paper too! #bioinformatics #biologicalDarkMatter #Gaia #tatta
03.03.2025 19:10 — 👍 11 🔁 3 💬 0 📌 1
(BioRxiv All) PepCentric Enables Fast Repository-Scale Proteogenomics Searches: Identifying novel peptides arising from alternative splicing, mutations, or non-canonical translations is a crucial yet challenging aspect of proteogenomics. We introduce… http://dlvr.it/TJFswF #BioRxiv #MassSpecRSS
01.03.2025 04:03 — 👍 0 🔁 1 💬 0 📌 0
Enhancing tandem MS sensitivity and peptide identification via ion pre-accumulation in an Orbitrap mass spectrometer www.biorxiv.org/cont...
---
#proteomics #prot-preprint

The Current Landscape of Plasma Proteomics: Technical Advances, Biological Insights, and Biomarker Discovery www.biorxiv.org/cont...
---
#proteomics #prot-preprint

VACANCY - We’re seeking a Proteomics Scientist to work alongside our experienced platform manager to help serve the growing needs of proteomics-based technology at the John Innes Centre (JIC).
www.jic.ac.uk/vacancies/pr...
Closing date - 2 March 2025
Contract - Full-time, indefinite

Identifying receptor kinase substrates using an 8,000 peptide kinase client library enriched for conserved phosphorylation sites #MCP #MassSpec www.mcponline.org/article/S153...
08.02.2025 08:25 — 👍 6 🔁 4 💬 0 📌 0