
Good read: www.mdpi.com/2571-9394/6/...
"Data-Centric Benchmarking of Neural Network Architectures for the Univariate Time Series Forecasting Task"
#timeseries #LSTM #realworlddata #neuralnetworks
Good read: www.mdpi.com/2571-9394/6/...
"Data-Centric Benchmarking of Neural Network Architectures for the Univariate Time Series Forecasting Task"
#timeseries #LSTM #realworlddata #neuralnetworks

The latest @livecomsjournal.bsky.social tutorial "Molecular Dynamics: From Basics to Application" by Vollmers, Chen et al is out now! doi.org/10.33011/liv...
It includes comprehensive MD tutorials in GROMACS, covering forcefields, thermodynamic ensembles, long-range electrostatics and much more!

#compchem #compbio Good read: Development of Coarse-Grained Lipid Force Fields Based on a Graph Neural Network pubs.acs.org/doi/10.1021/...
13.09.2025 07:07 — 👍 4 🔁 2 💬 0 📌 0
4️⃣ Featuring the fourth of our showcase projects
Upgrading GROMACS to handle billion-atom systems and enhancing I/O performance and precision, making the first-ever whole-cell simulation possible ➡️ bioexcel.eu/uw67
#MolecularDynamics #GROMACS #ComputationalBiology
Absolutely stunning.
08.09.2025 19:10 — 👍 1 🔁 0 💬 1 📌 0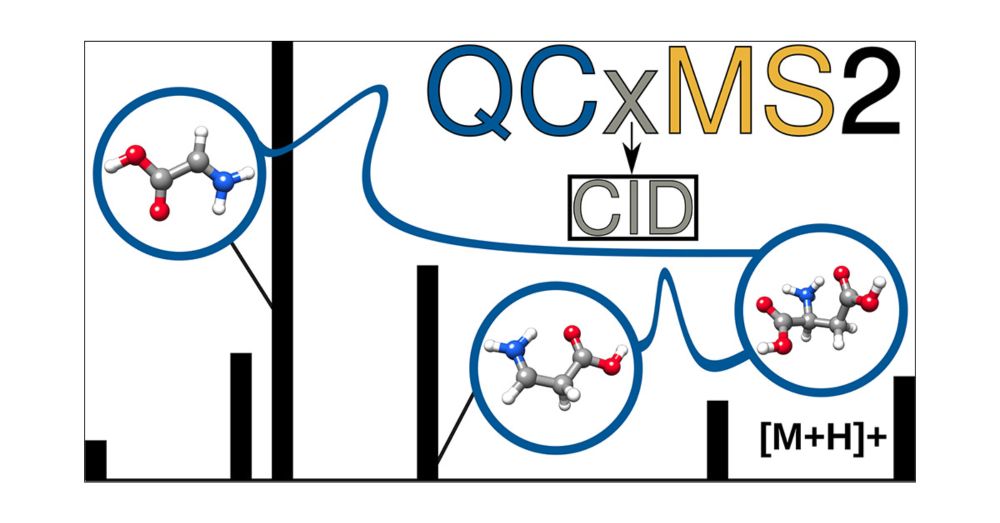
QCxMS2 can now also simulate CID mass spectra.
Just published in #JASMS : doi.org/10.1021/jasms.5c00234
Grateful to my coauthors Stefan Grimme @grimmelab.bsky.social & Marianne Engeser @unibonn.bsky.social - this is the last project of my PhD and completes my work on QCxMS2!
#MassSpec #compchem

How do proteins really fold? Our latest @pubs.acs.org JPCL paper with @saureli.bsky.social @valeriorizzi.bsky.social @mheritier.bsky.social unveils a new MD strategy to investigate it in atomistic resolution by focusing on water and side-chain interactions. check it out pubs.acs.org/doi/10.1021/...
08.09.2025 13:13 — 👍 5 🔁 2 💬 0 📌 0
Martini 3 Coarse-Grained Models for Carbon Nanomaterials | Journal of Chemical Theory and Computation pubs.acs.org/doi/full/10....
04.09.2025 10:07 — 👍 15 🔁 4 💬 0 📌 0
There we go... manuscript accepted in Nature.
From now on, I'm painting, playing games, and travelling 😀

Cleaning up my GitHub page. Most repositories are outdated, and the majority of work has been conducted on private company repositories. Nice picture of me and the dog, though 😅😍
github.com/acnash
Thanks so much. Sounds like I'm after MolecularNodes in Blender :-)
05.07.2025 13:14 — 👍 1 🔁 0 💬 1 📌 0
#compchem Our recent work "𝐒𝐡𝐨𝐫𝐭𝐜𝐮𝐭 𝐭𝐨 𝐜𝐡𝐞𝐦𝐢𝐜𝐚𝐥𝐥𝐲 𝐚𝐜𝐜𝐮𝐫𝐚𝐭𝐞 𝐪𝐮𝐚𝐧𝐭𝐮𝐦 𝐜𝐨𝐦𝐩𝐮𝐭𝐢𝐧𝐠 𝐯𝐢𝐚 𝐝𝐞𝐧𝐬𝐢𝐭𝐲-𝐛𝐚𝐬𝐞𝐝 𝐛𝐚𝐬𝐢𝐬-𝐬𝐞𝐭 𝐜𝐨𝐫𝐫𝐞𝐜𝐭𝐢𝐨𝐧 " has been selected in the following Nature collection ( #quantumcomputing for Quantum Chemistry section). www.nature.com/collections/...
28.06.2025 11:01 — 👍 12 🔁 4 💬 2 📌 0
Currently living here
github.com/acnash/gamd-...
#moleculardynamics #openmm #chemistry

I've adjusted the source code of Gaussian accelerated molecular dynamics (GAMD) with OpenMM (github.com/MiaoLab20/ga...) to accept periodic molecules, such as a sequence bonded to itself across the periodic boundary.
26.06.2025 08:48 — 👍 2 🔁 0 💬 1 📌 0And Python 2.7 on a different package. That's a sackable offense, surely ;-)
28.05.2025 11:53 — 👍 0 🔁 0 💬 0 📌 0
I'm exploring some software. I check out the dependencies... Perl, MatLab, BLAST, and DSSP.
This is going to break. I just know it.
#sciencesoftware
Our new preprint PharmacoForge: Pharmacophore Generation with Diffusion Models is out now! PharmacoForge quickly generates pharmacophores for a given protein pocket that identify key binding features and find useful compounds in a pharmacophore search. Check it out! 🧪 doi.org/10.26434/che...
27.05.2025 19:11 — 👍 21 🔁 9 💬 1 📌 0I've had to increase the font size used by the favourite IDE. Time stands still for no man.
27.05.2025 16:25 — 👍 0 🔁 0 💬 0 📌 0
New post: On the failure of rescoring in virtual screening.
Or why automated virtual screening and docking remains hard and why expertise remains essential. medchemash.substack.com/p/on-the-fai...
Thanks. MACE-MP-0 has zinc data, but further training would still be required to compensate for changes in the zinc coordination number in the binding pocket relative to my system. Thanks, this is a start.
21.05.2025 12:21 — 👍 1 🔁 0 💬 0 📌 0This looks great. Is it suitable for metalloproteases? Zinc protein binding centres, in particular?
21.05.2025 11:42 — 👍 0 🔁 0 💬 1 📌 0
Now out in @jacs.acspublications.org ! 🎉 : "MACE-OFF: Short-Range Transferable Machine Learning Force Fields for Organic Molecules" by Dávid Kovács, @jhmchem.bsky.social, & team:
pubs.acs.org/doi/10.1021/...
Agreed. I worked incredibly hard, but my career was derailed twice: Brexit and the Pandemic.
21.05.2025 08:18 — 👍 10 🔁 0 💬 2 📌 0#compchem
14.05.2025 21:59 — 👍 8 🔁 2 💬 0 📌 0
In today’s #good_practices #Journal_Club @clarakirkvold.bsky.social discusses the #FAIR #data principles and their implementations in #chemistry www.grynova-ccc.org/journal-club...
14.05.2025 17:45 — 👍 13 🔁 3 💬 0 📌 1
This is impressive! A huge QM structure of small molecules, ligands, biomolecules, etc., database.
Organisational skills must be at another level.
huggingface.co/facebook/OMo...
And the paper:
arxiv.org/abs/2505.08762

Can you keep up? I sometimes feel like I can't, but remember you're not alone. Just keep reading.
Some tips for performing meaningful and reproducible docking calculations.
journals.plos.org/ploscompbiol...
#docking #moleculardocking #liganddocking #compchem

ML/AI in the sciences is moving at an extraordinary pace. It's easy to feel left behind. Here's an excellent introduction to ML/AI concepts for the experimentalist and theoretician who is frantically reading to keep up.
#AI #ML #machinelearning #science
www.nature.com/articles/s41...
Yep! The absolute worst are applications that require your CV and a form dump of bits of your CV in a web portal. I've raised issues with admin staff about the replication of information. Sadly, it gets you know where.
12.05.2025 10:06 — 👍 1 🔁 0 💬 0 📌 0
Did you know I'm immortalized in plant form? There is a flower named after me.
Danum Anthony
www.dahliaworld.co.uk/dnamesv.htm#A
