
Try CAAT for yourself and let us know what you think.
github.com/prameshsharm...
Try CAAT for yourself and let us know what you think.
github.com/prameshsharm...

What about amino acids outside of these sparse patterns? AlphaFold is insensitive to their effects regardless of experimental ground truth in several cases. For example, XCL1 R23A R46A populates the red fold much more than wild type. XCL1 is blind to this difference.
05.01.2026 15:28 — 👍 1 🔁 0 💬 1 📌 0
Joey tested these mutations experimentally and found that they switch KaiB's fold.
05.01.2026 15:28 — 👍 1 🔁 0 💬 1 📌 0
We call this approach Conformational Attention Analysis Tool (CAAT). Applying CAAT to fold switcher KaiB revealed 4 important sites, 3 of which had been identified previously through hundreds of AlphaFold runs. CAAT requires just 2 AlphaFold passes. It suggested a new double mutant.
05.01.2026 15:28 — 👍 4 🔁 1 💬 1 📌 0
To understand the basis of AlphaFold's decision-making process, we retrieved the weights from its transformer model (Evoformer). Subtracting the weights between the two folds (gold and red) revealed AlphaFold's three "decision-making" amino acids (gold bars).
05.01.2026 15:28 — 👍 1 🔁 0 💬 1 📌 0
Madeleine found that both AlphaFold2 and AlphaFold3 oversimplify the mutations required for XCL1 to adopt the dimer fold. They predict the switch depends on just 3 amino acids, reducing AlphaFold's decision making process to a simple linear classifier.
05.01.2026 15:28 — 👍 1 🔁 0 💬 1 📌 0
Awhile back, Brian Volkman did beautiful work identifying complex sequence characteristics of fold switching in the human immunoprotein XCL1, which interconverts between monomeric chemokine (red) and beta-sheet dimer (gold) folds.
05.01.2026 15:28 — 👍 1 🔁 0 💬 1 📌 0
Check out Madeleine and Joey's latest work identifying specific amino acid patterns that AlphaFold uses to select protein conformations 🧵
www.biorxiv.org/content/10.6...
Small proteins can be more complex than they look!
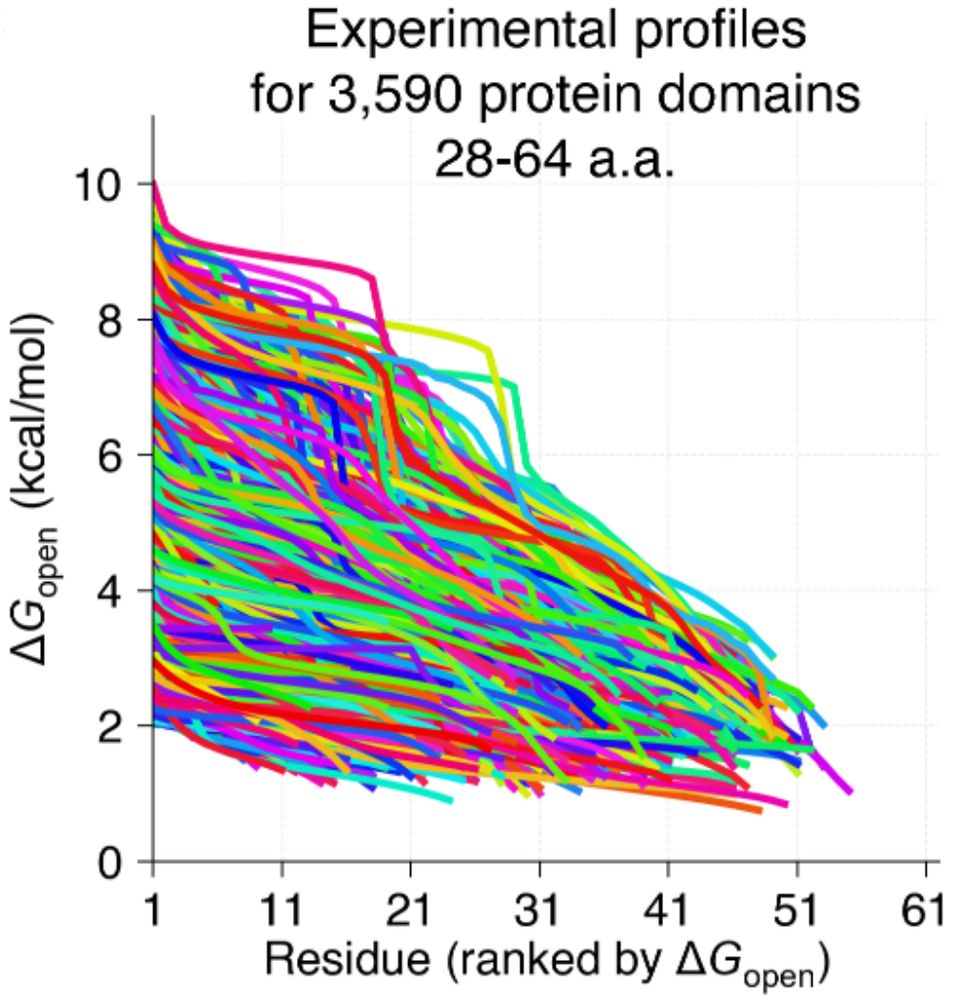
We know proteins fluctuate between different conformations- but by how much? How does it vary from protein to protein? Can highly stable domains have low stability segments? @ajrferrari.bsky.social experimentally tested >5,000 domains to find out!

🚨 New preprint alert! 🚨 How does SARS-CoV-2 use as much of its genome as possible to evade our immune response? Our latest study dissects how Orf9b, which is encoded in an alternate reading frame from the N(ucleocapsid) protein, can regulate interferon signaling.
www.biorxiv.org/content/10.1...
🧵👇

Our latest review is out in Current Opinion: Proteins with alternative folds reveal blind spots in AlphaFold-based protein structure prediction
www.sciencedirect.com/science/arti...
Sure. How would you suggest addressing it?
Doing my best to challenge the "because AI" argument! Science is all about explanations. If a neural network cannot be explained, seems to me it's not science. It's excellent engineering.
Agreed. I have never heard John Jumper or anyone else on the AF2 team overstate its capabilities. The confusion stems from other sources: misunderstandings of structural biology and overconfidence in AI.
14.12.2023 23:32 — 👍 5 🔁 0 💬 3 📌 0Thanks for the warm welcome 😀. It's good to be here.
14.12.2023 17:46 — 👍 1 🔁 0 💬 1 📌 0Agreed-- my lab uses AF2 all of the time (with caution) to generate hypotheses. Correct predictions do not imply learned folding physics.
14.12.2023 14:44 — 👍 5 🔁 0 💬 0 📌 0
These results lead me to believe that:
1). While AF2 builds great models, it does not equal experimental structure determination.
2). AF2 has not learned much protein folding physics. Rather, it has likely learned what good protein structures look like.

Together, these results suggest that AF2 has more to learn about protein energy landscapes. They also suggest that AF2 sometimes struggles to associate amino acid sequences with their correct conformations, as it did for BCCIP-alpha (correct fold on right):
14.12.2023 11:53 — 👍 2 🔁 1 💬 1 📌 0
Further, AF2 and AF-cluster failed to predict new fold switchers discovered after AF2.3.1 was trained:
14.12.2023 11:52 — 👍 4 🔁 0 💬 1 📌 0
AF2 based methods performed worse when predicting fold switchers outside of the training set. For instance, AF-cluster predictions could not distinguish between sequence-diverse fold-switching and single-folding RfaH homologs, and all helical predictions had low confidence.
14.12.2023 11:52 — 👍 3 🔁 0 💬 1 📌 0
Consistently, AF2's structure module--hypothesized to have learned folding physics--could not discriminate between low and high energy conformations of fold switchers. (TM-scores on x- and y- axes).
14.12.2023 11:51 — 👍 3 🔁 0 💬 1 📌 0
AF2 confidence metrics selected against diverse experimentally observed conformations, particularly alternative folds (p < 8.1*10-4, one-sided binomial test), in favor of experimentally unobserved conformations
14.12.2023 11:50 — 👍 4 🔁 0 💬 1 📌 0
We tested 5 AF2-based methods on 93 fold-switching proteins likely in AF2’s training set, >280,000 predictions total. Each method predicted <20% of fold switchers and favored experimentally unobserved folds (other) over experimentally observed alternative folds (Fold2).
14.12.2023 11:50 — 👍 5 🔁 0 💬 1 📌 0
Our latest @bioRxiv manuscript reports an assessment of >500,000 AF2-based predictions of fold-switching proteins, which assume two distinct stable structures. These predictions fail to reflect observed energetics of fold switching 🧵 www.biorxiv.org/content/10.1...
14.12.2023 11:49 — 👍 32 🔁 9 💬 1 📌 0