There's lot more interesting things we looked at:
- We find some credible transmission links in India
- We also see some kind of (likely) reduction IS-mediated funny business going on in the linear plasmid.
Checkout the preprint for all the details.
05.03.2026 05:21 —
👍 0
🔁 0
💬 0
📌 0

We deemed this transposon Tn8026. We did some global screening and found Tn8026 in a variety of countries, with the earliest evidence being Norway in 2012. We also found it in 2 E. gallinarum isolates from S. Korea. PLUS it was also in the chromosome of one of our isolates!
05.03.2026 05:21 —
👍 0
🔁 0
💬 1
📌 0
To add to the intrigue, the linezolid resistance mechanism, a gene called poxtA-Ef, was located on this linear plasmid, along with Tn1546, which carries the vancomycin resistance gene cluster.
Upon further inspection, we realised poxtA-Ef was in what turns out to be an uncharacterised transposon
05.03.2026 05:21 —
👍 0
🔁 0
💬 1
📌 0
Turns out most of these isolates had a LINEAR plasmid. Really showing my inexperience here as I did not know that was a thing.
After doing some more reading I found that Jia Beh from the Doherty in Melbourne, Aus. had also found a linear plasmid in some LREs (as had a couple of others globally)
05.03.2026 05:21 —
👍 0
🔁 0
💬 1
📌 0
The dataset was linezolid resistant Enterococcus (LRE), which are very concerning pathogens that are resistant to nearly everything.
We sequenced all these on ONT and I started by making assemblies. First shout out to @rrwick.bsky.social for the beautiful piece of software that is Autocycler!
05.03.2026 05:21 —
👍 0
🔁 0
💬 1
📌 0
Does anyone else think they are seeing post-acceptance editorial changes at proof stage which are error-prone and probably due to adoption of AI?
02.12.2025 10:29 —
👍 4
🔁 2
💬 4
📌 1

GitHub - mbhall88/nohuman: Remove human reads from a sequencing run
Remove human reads from a sequencing run. Contribute to mbhall88/nohuman development by creating an account on GitHub.
So nohuman now ships an unmasked HPRC.r2 DB by default, with optional dataset selection.
If you’ve used nohuman before, I highly recommend updating to v0.5.0 and re-downloading the new DB.
Repo: github.com/mbhall88/nohuman
Keep your metagenomes clean 🧹🧬
20.11.2025 06:50 —
👍 1
🔁 1
💬 0
📌 0

At the same time, I realised the Human Pnagenome Reference Consortium had made a second release of genomes.
So I rebuilt release 1 without masking, and added a release 2 database with no masking. The improvement in detection accuracy was substantial:
20.11.2025 06:50 —
👍 0
🔁 0
💬 1
📌 0
🚨 Update to nohuman 🚨
While testing against the standard Kraken DB, I noticed Kraken was detecting far more human reads than nohuman. I realised Kraken masks low-complexity regions by default during DB construction and that setting had been left on in nohuman, leading to missing human reads.
20.11.2025 06:50 —
👍 2
🔁 1
💬 1
📌 0
Stars are level of p value (description is in the figure caption in the paper)
07.11.2025 19:43 —
👍 1
🔁 0
💬 0
📌 0
True.
Thanks for the great questions and discussion
07.11.2025 11:07 —
👍 1
🔁 0
💬 1
📌 0
Correct. Yeah I guess mash on a random subset should perform similarly. Haven’t looked at that though.
07.11.2025 11:05 —
👍 1
🔁 0
💬 0
📌 0
It’s a decent sample size at 3000. But I guess more would always be better. I wanted to use refseq genomes which has long read data to be as sure as possible about the true size
There is likely inherent biases though based on error rates in reads for the kmer based methods
07.11.2025 11:04 —
👍 0
🔁 0
💬 1
📌 0
- Overlaps are pairwise alignment with minima2 (FFI)
-Thanks!
- See other thread where I have answered this
07.11.2025 11:01 —
👍 2
🔁 0
💬 0
📌 0
I just used mash v2.3. The supplement has an exploration of the best parameters to use for mash to estimate genome size. Mash was the fastest tool though.
07.11.2025 10:58 —
👍 0
🔁 0
💬 0
📌 0
the bars are pair wise statistical comparisons. I only show the significant ones so as not to over clutter the plot
07.11.2025 10:52 —
👍 1
🔁 0
💬 1
📌 0
And lastly, a HUGE thank you to @lachlanjmc.bsky.social for a lot of the methodological heavy lifting when we were coming up with the idea
07.11.2025 03:21 —
👍 0
🔁 0
💬 0
📌 0
You might remember the preprint from late last year... Reviews/Publication were delayed while I was on parental leave. We extended validation to include H. sapiens, which lead to smarter handling of contained overlaps in repetitive genomes. Big shout-out to Chenxi Zhou for leading that part
07.11.2025 03:18 —
👍 0
🔁 0
💬 1
📌 0

However, the computational resource usage (runtime/memory) of LRGE was MUCH better than assembling
07.11.2025 03:18 —
👍 0
🔁 0
💬 1
📌 0

We benchmarked >3,000 bacterial genomes and found that LRGE (our method) achieves significantly better accuracy than k-mer-based methods like Mash and GenomeScope and performs on par with full genome assembly (Raven)
07.11.2025 03:18 —
👍 0
🔁 0
💬 2
📌 0
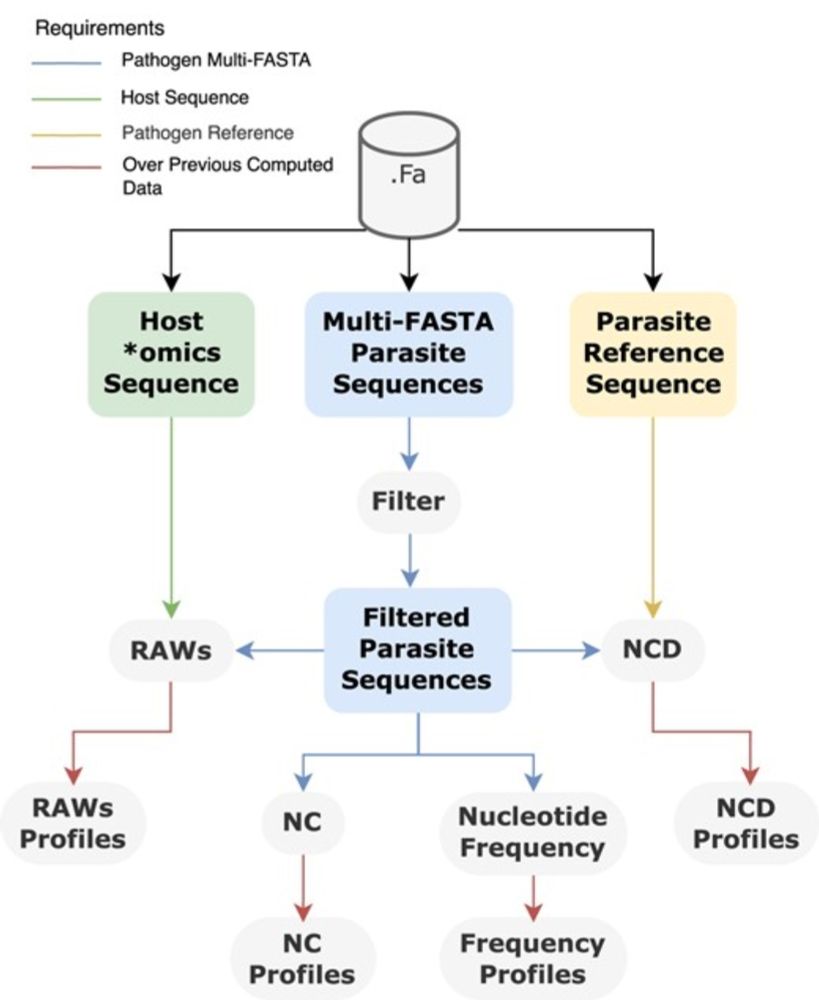
AltaiR: a C toolkit for alignment-free and temporal analysis of multi-FASTA data
AbstractBackground. Most viral genome sequences generated during the latest pandemic have presented new challenges for computational analysis. Analyzing mi
New from @dgpratas.bsky.social et al. for analyzing multiple sequences in multi-FASTA format using alignment-free methodologies. Scalable to millions of sequences for pandemic research and more
AltaiR: a C toolkit for alignment-free and temporal analysis of multi-FASTA data doi.org/10.1093/giga...
12.12.2024 10:28 —
👍 4
🔁 4
💬 0
📌 0

How the Web of Science takes a step back
<p>The Web of Science, a major commercial indexing service of scientific journals operated by Clarivate, recently decided to remove eLife from its Science Citation Index Expanded (SCIE). eLife will on...
“Clarivate’s decision rewards journals for continuing the unhelpful practice of keeping peer review information hidden and unintentionally presenting incomplete and inadequate studies as sound science and punishes those journals that are more transparent.” 👏🙌
www.coalition-s.org/blog/how-the...
03.12.2024 09:49 —
👍 3
🔁 0
💬 0
📌 0
The DOI URL doesn't seem to be working for the preprint currently. You can find it here: www.biorxiv.org/content/10.1...
03.12.2024 04:02 —
👍 0
🔁 0
💬 0
📌 0
GitHub - mbhall88/lrge: Genome size estimation from long read overlaps
Genome size estimation from long read overlaps. Contribute to mbhall88/lrge development by creating an account on GitHub.
8/ Try it out!
LRGE is open-source and ready to integrate into your workflows as a Rust library or CLI application. Whether you’re on a high-performance cluster or a basic laptop, LRGE delivers fast and reliable genome size estimates. Get it here: github.com/mbhall88/lrge
03.12.2024 01:38 —
👍 3
🔁 0
💬 0
📌 0
7/ We validated LRGE on 3370 long read bacterial datasets which have associated high-quality RefSeq assemblies 🦠. We also confirmed it generalises to eukaryote organisms 🪰🌱🍞
03.12.2024 01:38 —
👍 0
🔁 0
💬 1
📌 0
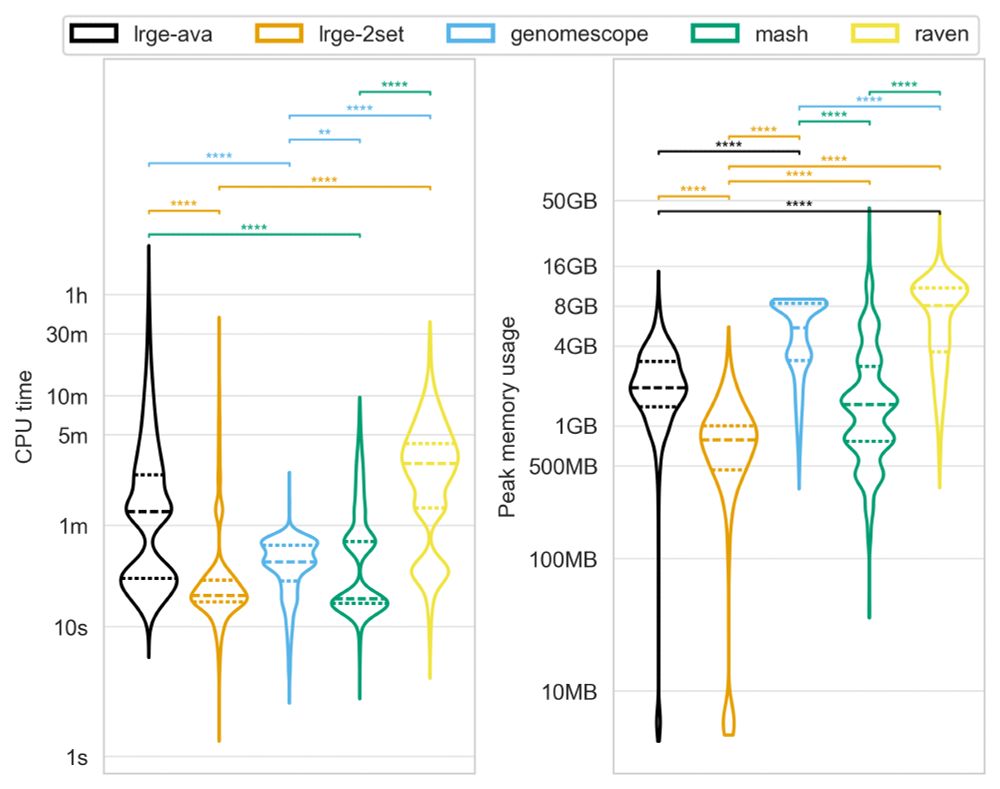
6/ And it’s efficient! ⚡
LRGE uses significantly less CPU and memory than traditional approaches, making it ideal for both high-performance clusters and resource-limited environments.
03.12.2024 01:38 —
👍 0
🔁 0
💬 1
📌 0




