
Chemical Space Exploration with Artificial “Mindless” Molecules
We introduce MindlessGen, a Python-based generator for creating chemically diverse, “mindless” molecules through random atomic placement and subsequent geometry optimization. Using this framework, we ...
🚀 JCIM: "Chemical Space Exploration with Artificial Mindless Molecules"
We present MindlessGen, an open-source tool for generating chemically diverse "mindless" molecules, and the MB2061 benchmark set with high-level reference data to test methods on unconventional systems.
doi.org/10.1021/acs....
03.09.2025 11:01 — 👍 22 🔁 3 💬 1 📌 0

We are delighted to announce that our perspective article, “Steering towards safe self-driving laboratories (SDLs),” has been accepted for publication in Nature Reviews Chemistry.
Link: www.nature.com/articles/s41...
18.08.2025 17:46 — 👍 15 🔁 7 💬 1 📌 1


Headed to Accelerate 2025? You cannot miss the presentations from The Matter Lab. We've got your back--here's your ultimate cheat sheet so you don't miss a thing.
#Accelerate2025 #AIinScience
[1/2]
08.08.2025 18:28 — 👍 5 🔁 3 💬 1 📌 1
ORCA as External Optimizer - ORCA 6.1 TUTORIALS
You can now use g-xTB @grimmelab.bsky.social with ORCA via the ExtOpt feature! Check out our new tutorial and learn how to use it in GOAT, NEB-TS and more.
www.faccts.de/docs/orca/6....
#ORCAqc #FACCTs #gxTB #CompChem #QuantumChem
26.06.2025 08:32 — 👍 39 🔁 9 💬 2 📌 1
We are working in this direction. However, analytical expressions for the nuclear gradient (or at least their implementation) get much more complicated in ab-initio methods, when using an atom-in-molecule-adaptive basis set.
24.06.2025 20:13 — 👍 1 🔁 0 💬 0 📌 0
Excited states-support is a feature that will also be available with g-xTB in the future (in the final implementation). Stay tuned! :)
24.06.2025 14:37 — 👍 5 🔁 0 💬 0 📌 0
📢 Update on our g-xTB release published on ChemRxiv:
We’ve uploaded a Linux executable of the current development version of g-xTB on GitHub, along with a simple usage guide:
🔗 github.com/grimme-lab/g...
⚠️ Please note:
This is a preliminary release — currently Linux-only, using numerical gradients.
24.06.2025 13:07 — 👍 0 🔁 0 💬 0 📌 0
g-xTB excels in areas where SQM and even DFT often struggle:
✅ Transition-metal thermochemistry
✅ Spin-state energies
✅ Orbital energy gaps
✅ Reaction barriers
And all that at a fraction of DFT cost.
24.06.2025 07:31 — 👍 3 🔁 0 💬 1 📌 0
g-xTB is built to replace GFN2-xTB in all applications.
It cuts MAEs by half, improves SCF convergence, and even beats B3LYP-D4 for reaction barriers — all with just 30–50% more computational cost than GFN2-xTB.
24.06.2025 07:31 — 👍 4 🔁 1 💬 2 📌 0

Some key highlights of g-xTB — our first general-purpose xTB method delivering DFT accuracy at SQM speed.
It tackles not only geometries, frequencies, and NCIs ("GFN"), but also strong thermochemistry and electronic properties with unprecedented accuracy for a semiempirical method.
🔗 #compchem
24.06.2025 07:31 — 👍 4 🔁 1 💬 1 📌 0
Two of them are at #WATOC2025 this week and ready to share all the details about the method you’ve been waiting for:
📍 @thfroitzheim.bsky.social — Thursday, Session B1, 9:20 AM
📍 S. Grimme — Thursday, Session A2, 10:20 AM
Don’t miss it!
24.06.2025 07:31 — 👍 5 🔁 1 💬 1 📌 0
Big thanks to my amazing co-workers: @thfroitzheim.bsky.social, Stefan Grimme, and Andreas Hansen! 🎉
24.06.2025 07:31 — 👍 3 🔁 0 💬 1 📌 0
I see it more as a form of art 😂
23.06.2025 23:17 — 👍 0 🔁 0 💬 0 📌 0
I immediately loved the optical appearance of the molecules in this figure when I created it. 😂 But yeah, "unhinged" is very accurate! That's exactly what we wanted. 🤓
23.06.2025 14:52 — 👍 1 🔁 0 💬 1 📌 0

Chemical Space Exploration with Artificial ”Mindless” Molecules
We introduce MindlessGen, a Python-based generator for creating chemically diverse, “mindless” molecules through random atomic placement and subsequent geometry optimization. Using this framework, we constructed the MB2061 benchmark set, containing 2061 molecules with high-level PNO-LCCSD(T)-F12 reference data for dissociation reactions. This set provides a challenging benchmark for testing, validation, and training of density functional approximations (DFAs), semiempirical methods, force fields, and machine learning potentials using molecular structures beyond the conventional chemical space. For DFAs, we initially hypothesized that highly parameterized functionals might perform poorly on this set. However, no consistent relationship between fitting strategy and accuracy was observed. A clear Jacob’s ladder trend emerges, with ωB97X-2 achieving the lowest mean absolute error (MAE) of 8.4 kcal·mol−1 and r²SCAN-3c offering a robust cost-efficient alternative (19.6 kcal·mol−1). Furthermore, we discuss the performance of selected semiempirical methods and contemporary machine learned interatomic potentials.
#RobSelects preprint of the week #ChemRxiv: Benchmarking density functional approximations with a systematic set of randomly generated molecules. #compchem https://doi.org/10.26434/chemrxiv-2025-rdsd0
18.06.2025 08:37 — 👍 5 🔁 4 💬 1 📌 0
Say Hello to the Bannwarth group at Bluesky and give them a follow for great science! 👋 @bannwarthlab.bsky.social 🚀🔬
14.02.2025 18:25 — 👍 1 🔁 0 💬 0 📌 0

Thank you for your question! While an energy expression in the context of density-corrected DFT can still be conceptually very inspiring, we are currently working on a “real” xTB successor, called g-xTB.
This plot about the accuracy of the barrier heights compared to DFT gives a good impression. 💡
30.01.2025 09:53 — 👍 3 🔁 1 💬 0 📌 0
Thank you for your question! While an energy expression in the context of density-corrected DFT can still be conceptually very inspiring, we are currently working on a “real” xTB successor, called g-xTB.
This plot about the accuracy of the barrier heights compared to DFT gives a good impression. 💡
30.01.2025 09:53 — 👍 3 🔁 1 💬 0 📌 0
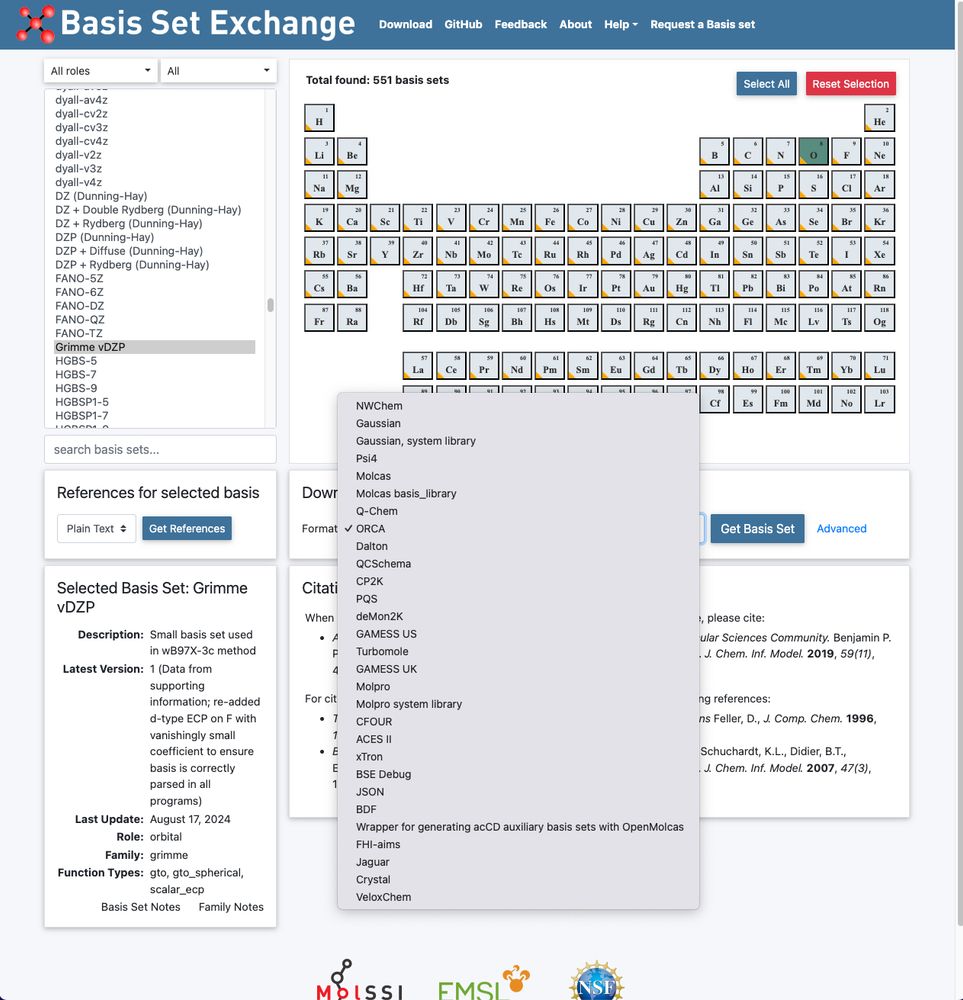
Our vDZP basis set utilized in the ⍵B97X-3c composite DFT method is now also available via www.basissetexchange.org (API-based: github.com/MolSSI-BSE/b...). 🎉
Many thanks to @Susi Lehtola & coworkers for jointly providing it there!
29.01.2025 16:02 — 👍 29 🔁 7 💬 2 📌 0
This is a question I can only answer with a certain bias, as we are actively developing xTB and related tight-binding methods (which have their roots in DFTB). From this point of view, I would answer “No, xTB has become the standard, at least for molecular systems with less than about 2000 atoms.” 🤓
23.01.2025 18:21 — 👍 1 🔁 0 💬 0 📌 0
2. See this answer: bsky.app/profile/mrcl...
Thus, I consider models that have a built-in quantum chemical foundation in them as semiempirical (in the sense of theoretical chemistry/quantum chemistry methods).
23.01.2025 10:56 — 👍 1 🔁 0 💬 0 📌 0
1. Good suggestion. I wasn't sure if I should consider it semiempirical, since in a sense it could also be a precursor to KS-DFT.
23.01.2025 10:55 — 👍 1 🔁 0 💬 0 📌 0
Personally, I would consider the idea of machine-learning potentials or force fields as empirical (not semiempirical), since they derive their behavior mainly from the emulation of reference data (e.g. DFT) and carry only a limited amount of physics (e.g. no quantized energy levels).
23.01.2025 10:52 — 👍 2 🔁 0 💬 0 📌 1
Happy to announce the release of @avogadro.cc 1.100 (not quite 2.0 yet) with a pile of new features, bug fixes, etc.
Can't fit the whole release notes here, but more on the forum:
discuss.avogadro.cc/t/avogadro-1...
22.01.2025 14:13 — 👍 24 🔁 5 💬 1 📌 0
Thanks for the remark, I will add his name to the NDO part.
22.01.2025 12:53 — 👍 1 🔁 0 💬 0 📌 0

I have created a timeline of semiempirical methods in quantum chemistry. Any thoughts, suggestions, or remarks on it? 💡 Have we missed anything?
22.01.2025 12:22 — 👍 19 🔁 2 💬 4 📌 1
Principal Research Scientist at NVIDIA | Former Physicist | Deep Generative Learning | https://karstenkreis.github.io/
Opinions are my own.
We design functional molecules and materials for catalysis and sustainable energy technologies using quantum simulations.
Amir Karton, Associate Dean of Research, University of New England, Armidale, Australia
An international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry.
🌐 Website rsc.li/PCCP
computational chemist @ RUB | weightlifting&BJJ for fun, yoga for recovery | reader, but not an avid one
Dad | Aspiring Scientist | Theoretical Chemist at Leipzig University | Some Other Things | patrickmelix.de
Student-run twitter account for the Hartwig Group at UC Berkeley. 🧪
https://hartwig.cchem.berkeley.edu/
PhD candidate in the Hartwig Group.
Working on combining transition-metal catalysis with data science, machine learning, and high-throughput experimentation to accelerate discovery in synthetic chemistry.
PhD | Quantum Chemist | meet.abaldo.link
Flagship journal for @rsc.org, open access with no publication fee, all topics in chemistry. chemicalscience-rsc@rsc.org
Celebrating our 15th year in 2025! 🌐 Website: rsc.li/chemscience
PhD Student in Machine Learning for Chemistry in the @westermayrgroup.bsky.social @unileipzig.bsky.social
PhD at Leipzig University and TU Berlin working on machine learning for quantum chemistry
Research group of Junior Professor Julia Westermayr
Wilhelm-Ostwald-Institute for Physical and Theoretical Chemistry
Leipzig University
We are a computational research group at Newcastle University led by #UKRIFLF Dr Danny Cole specialising in atomistic simulations in medicinal chemistry and biology https://blogs.ncl.ac.uk/danielcole/
IR Scholar. Universität der Bundeswehr München. Private account
Nachrichten, Analysen, Kommentare, Videos, Podcasts: Mehr als 500 SPIEGEL-Journalistinnen und Journalisten decken auf, setzen Themen und sorgen für Kontext.
https://www.spiegel.de/
























