
🧬 New preprint alert! After years of collaborative work across 52 datasets we are presenting eQTLGen phase 2: a genome-wide eQTL meta-analysis covering 43,301 blood samples: www.medrxiv.org/content/10.6... (1/8)
06.02.2026 10:14 — 👍 36 🔁 21 💬 1 📌 4
@magiduck.bsky.social
PhD student within the Functional Genomics group (Franke lab), University Medical Centre Groningen. Interested in gene networks, sequence-based models and non-coding somatic mutations
🧬 New preprint alert! After years of collaborative work across 52 datasets we are presenting eQTLGen phase 2: a genome-wide eQTL meta-analysis covering 43,301 blood samples: www.medrxiv.org/content/10.6... (1/8)
06.02.2026 10:14 — 👍 36 🔁 21 💬 1 📌 4
Tomorrow at 14.30 I'll present my poster on the Identification of (ultra-)rare functional promoter mutations in cancer using sequence-based deep learning models at #ashg. Swing by poster Board 9050F if you want to chat or know more! #ashg2025
16.10.2025 18:20 — 👍 4 🔁 0 💬 0 📌 0
We are excited to share GPN-Star, a cost-effective, biologically grounded genomic language modeling framework that achieves state-of-the-art performance across a wide range of variant effect prediction tasks relevant to human genetics.
www.biorxiv.org/content/10.1...
(1/n)
Excited for a major milestone in our efforts to map enhancers and interpret variants in the human genome:
The E2G Portal! e2g.stanford.edu
This collates our predictions of enhancer-gene regulatory interactions across >1,600 cell types and tissues.
Uses cases 👇
1/

An assessment of DNA language models concludes:
◼️ They do not offer compelling gains over baseline models
Their performance is inconsistent and requires much more compute.
arxiv.org/abs/2412.05430
I'm only partially kidding cuz he definitely is forcing me to modify some of my slides 😂. But I did want to express some thoughts on the lightweight specialized single task (ST) models vs kitchensink multi task (MT) model paradigms a bit. 2/
26.06.2025 06:29 — 👍 3 🔁 1 💬 1 📌 0This a really exciting leap forward for genomic sequence to activity gene regulation models. It is a genuine improvement over pretty much all SOTA models spanning a wide range of regulatory, transcriptional and post-transcriptional processes. 1/
25.06.2025 16:18 — 👍 73 🔁 20 💬 2 📌 2One thing that really bothers me with the new "virtual cell" terminology is that it is currently largely focused on a very narrow definition of models that can predict effects of trans perturbations (gene dosage, drugs etc) on gene expression. 1/
28.06.2025 10:38 — 👍 104 🔁 29 💬 1 📌 0
1/ DNA sequence models like Borzoi predict gene expression and variant effects across tissues — but how can someone adapt the model to a custom experiment? @drkbio.bsky.social, Johannes Linder and I propose a solution via parameter-efficient fine-tuning (PEFT).
www.biorxiv.org/content/10.1...
Many of you enjoy our sequence-based model of single-cell RNA and ATAC data scooby... Don't miss Laura Marten's talk at the upcoming Kipoi seminar about it this Wed!
@lauradmartens.bsky.social @johahi.bsky.social @kipoizoo.bsky.social
Last preprint version:
www.biorxiv.org/content/10.1...
We studied non-coding somatic mutations in WGS data of 24,000 cancer patients. We employed sequence-based models to identify promotors, enriched for somatic variants that are predicted to affect expression. A great collaboration with the teams of Emile Voest, Bas van Steensel and Jeroen de Ridder!
07.05.2025 08:03 — 👍 2 🔁 1 💬 0 📌 0Identification of (ultra-)rare functional promoter mutations in cancer using sequence-based deep learning models https://www.medrxiv.org/content/10.1101/2025.05.06.25327057v1
07.05.2025 03:40 — 👍 1 🔁 1 💬 0 📌 0This paper is also supported by resources from intOGen (@nlbigas.bsky.social ) and Cosmic Cancer Gene Census. (16/16)
07.05.2025 07:04 — 👍 1 🔁 0 💬 0 📌 0Also big thanks to the authors of PARM (@luciabarmar.bsky.social et al) and Borzoi (Johannes Linder and @drkbio.bsky.social et al) for sharing their models. (15/16)
07.05.2025 07:04 — 👍 2 🔁 0 💬 1 📌 0Thanks to all the other co-authors, Lucía Barbadilla-Martínez, Minh Chau Luong Boi, Harm-Jan Westra, Noud H.M. Klaassen, Vinícius H. Franceschini-Santos, Miguel Parra-Martínez, Jeroen de Ridder, Bas van Steensel, Emile Voest and Lude H. Franke for the collaboration! (14/16)
07.05.2025 07:04 — 👍 1 🔁 0 💬 1 📌 0Many thanks to all the co-authors (in particular the close collaboration with Carlos Urzúa-Traslaviña) for their input, discussions, collaboration and reviews! (13/16)
07.05.2025 07:04 — 👍 1 🔁 0 💬 1 📌 0Our results suggests that the promoter regions of many genes contain non-coding somatic mutations that contribute to cancer development. This is substantially more than what has been reported previously. (12/16)
07.05.2025 07:04 — 👍 1 🔁 0 💬 1 📌 0
These genes are enriched for being involved in the cell-cycle, being known cancer driver genes and being more evolutionary constrained. (11/16)
07.05.2025 07:04 — 👍 1 🔁 0 💬 1 📌 0A meta-analysis across all 24,529 samples reveals 492 regions to be significantly enriched for non-coding somatic mutations with a predicted functional impact. (10/16)
07.05.2025 07:04 — 👍 1 🔁 0 💬 1 📌 0
We identify nine high-confident regions (including the well-known TERT, TP53 and PMS2 genes) where in three independent cohorts we find significant enrichment of non-coding mutations and where we can also confirm that these mutations show actual effects on gene expression levels. (9/16)
07.05.2025 07:04 — 👍 2 🔁 0 💬 1 📌 0We identified 492 genes where this is the case, indicating that somatic non-coding mutations play a substantial role in causing cancer. This is more than earlier studies which had to rely on smaller sample sizes and mutational background models that can be quite challenging to apply. (8/16)
07.05.2025 07:04 — 👍 1 🔁 0 💬 1 📌 0
We subsequently performed a burden analysis to identify a comprehensive set of genes that are enriched for containing non-coding somatic mutations that either preferentially activate or preferentially repress gene expression. (7/16)
07.05.2025 07:04 — 👍 2 🔁 0 💬 1 📌 0
Here we used a novel strategy to do this in WGS data of 24,529 cancer patients: we used sequence-based models to predict the transcriptional consequences of these mutations. (6/16)
07.05.2025 07:04 — 👍 1 🔁 0 💬 1 📌 0
This is because no algorithms yet existed to distinguish functional from irrelevant non-coding mutations. (5/16)
07.05.2025 07:04 — 👍 1 🔁 0 💬 1 📌 0This was surprising, considering that 97% of all mutations occur in non-coding regions. However, these statistical approaches had to lump together all mutations, irrespective of whether these mutations may alter gene regulation or not. (4/16)
07.05.2025 07:04 — 👍 1 🔁 0 💬 1 📌 0However, previous statistical approaches to find non-coding “driver” mutations in large cancer genome datasets have so far revealed a few genes that showed mutation frequencies in their regulatory elements that were higher than expected by chance. (3/16)
07.05.2025 07:04 — 👍 1 🔁 0 💬 1 📌 0With the availability of large amounts of whole-genome sequence (WGS) data in tumor biopsies, a major opportunity now exists to determine which somatic non-coding mutations contribute to cancer and which do not. (2/16)
07.05.2025 07:04 — 👍 1 🔁 0 💬 1 📌 0
We are excited to share our manuscript “Identification of (ultra-)rare functional promoter mutations in cancer using sequence-based deep learning models” (www.medrxiv.org/content/10.1...). (1/16) 🧵
07.05.2025 07:04 — 👍 6 🔁 1 💬 1 📌 1As someone who has reported on AI for 7 years and covered China tech as well, I think the biggest lesson to be drawn from DeepSeek is the huge cracks it illustrates with the current dominant paradigm of AI development. A long thread. 1/
27.01.2025 14:12 — 👍 6164 🔁 2360 💬 211 📌 722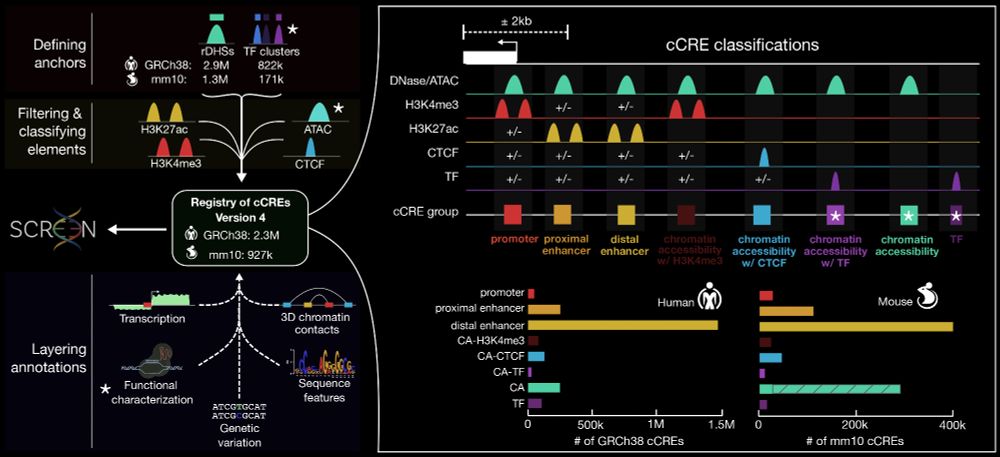
Schematic overview of the ENCODE 4 project's framework for identifying and classifying candidate cis-regulatory elements (cCREs). The process begins by defining regulatory anchors, such as DNase hypersensitive sites (rDHSs) and transcription factor (TF) clusters, which are filtered and classified using histone marks and other features. Elements are classified into functional categories, including promoters, proximal and distal enhancers, and CTCF-bound sites. The resulting registry includes millions of cCREs for human (GRCh38) and mouse (mm10), depicted alongside their classifications. Additional annotations, such as transcription activity, chromatin structure, genetic variation, and sequence motifs, are integrated for functional characterization. Image modified from https://www.biorxiv.org/content/10.1101/2024.12.26.629296v1.full
Now available as preprint:
The ENCODE 4 expanded registry of regulatory elements
- 2.35M 🧍 human cCREs
- 927k 🐭 mouse cCREs
www.biorxiv.org/content/10.1...
Led by @moorejille.bsky.social, this preprint summarizes data and analyses generated by hundreds of contributors across ENCODE 4