




A dataset of 40 million protein families and an autoregressive model of protein families. Great to see other protein Atlases popping up after Dayhoff!!
@judewells.bsky.social @dmmiller597.bsky.social
www.biorxiv.org/content/10.6...
A dataset of 40 million protein families and an autoregressive model of protein families. Great to see other protein Atlases popping up after Dayhoff!!
@judewells.bsky.social @dmmiller597.bsky.social
www.biorxiv.org/content/10.6...
Built by CATH, TÜM and NVIDIA, ProFam-1 is our new open-source protein family language model (pfLM) designed to generate functional protein variants and predict fitness using in-context example sequences.
22.12.2025 14:32 — 👍 11 🔁 5 💬 1 📌 1Finally @adaptyv.bio today is my birthday, so if you could please test my designs, I would be very grateful :)
01.12.2025 15:44 — 👍 2 🔁 0 💬 0 📌 0Assuming that the loop identity alone is sufficient for binding is probably an oversimplification, it might require allosteric changes for binding or other parts of the anitbody interface may also be essential. Perhaps it would have been wise to try and tune more the interface sites on the scafolds
01.12.2025 15:44 — 👍 1 🔁 0 💬 1 📌 0If I had more time, I would have tried out more scaffolds & different splicing strategies. Normally, I would try and optimise ipsae, MPNNsol likelihood, aim for consensus structures from chai, boltz, alphafold. But also it's fun to say that I did one-shot design in Microsoft Word
01.12.2025 15:44 — 👍 0 🔁 0 💬 1 📌 0interestingly, the Boltz predictions on the Adaptyv Bio site are different to my colabfold predicted structures. (left to right) PDB of antibody, colabdold on my scFv & boltz on my scFv on the right.
01.12.2025 15:44 — 👍 1 🔁 0 💬 1 📌 0I tried one other scaffold which is this APPI kunitz domain (orange PDB 3l3t) but the splice is incompatible with this cystein bridge + hydrogen bond network. AlphaFold predicts that splicing breaks the fold: and the loop doesn't end up in the right place for binding, not hopeful for that one
01.12.2025 15:44 — 👍 1 🔁 1 💬 1 📌 0Here’s the original antibody 1e5 on the left and my designed scvf on the right. AlphaFold prediction is consistent with the original antibody so that means: send it.
01.12.2025 15:44 — 👍 1 🔁 1 💬 1 📌 0First I found a scFv in the pdb that used g4s (PDB 2GHW left) whose structure spanned 33Å with a 16-res linker. the gap to fuse the two 1E5 chains is 26Å (right) but it needs to take a curved path: hopefully, 16-res is still sufficient
01.12.2025 15:44 — 👍 0 🔁 0 💬 1 📌 0Strategy 2: try and turn the 1E5 antibody into a Single-chain variable fragment (scFv) by getting rid of the two non-binding domains and fusing the 2 remaining binding domains into a single chain. With glycine(x4)-serine (g4s) linker.
01.12.2025 15:44 — 👍 0 🔁 0 💬 1 📌 0We start with the designed scaffold protein called Adhiron (left PDB 4N6T) and splice in the 13 residue loop from the antibody. Colabfold structure on the right shows spliced residues (red) binding in the same location as the antibody: send it.
01.12.2025 15:44 — 👍 0 🔁 0 💬 1 📌 0The antibody is too big ~450 residues and it’s 2 chains instead of 1. So strategy1 : take the burried loop from the antibody and splice into a single domain scaffold protein.
01.12.2025 15:44 — 👍 1 🔁 0 💬 1 📌 0Step 1 find known binders for this viral protein: we find an antibody called 1E5 (PDB 8XC4 left). We can align the viral-human complex with the viral antibody complex (right) and see that both the human receptor and antibody have a coil burried in the virus central cavity.
01.12.2025 15:44 — 👍 0 🔁 0 💬 1 📌 0The target protein is Nipah glyco-protein G (left), and in complex with cognate human receptor ephrinB2 shown in white on the right. (PDB 2VSM)
01.12.2025 15:44 — 👍 0 🔁 0 💬 1 📌 0Ok let’s go @adaptyv.bio binder design competition: this time designing proteins to neutralise the Nipah virus. Lots of great de novo ML binder design tools out there now, but this year I’m submitting an entry from TEAM HUMAN, seeing if pure rational design can win against the machines.
01.12.2025 15:44 — 👍 2 🔁 0 💬 1 📌 0
🚀 As first official act, we are hiring! 🎓
We’re looking for a PhD student to work at the interface of computational biophysics, machine learning & human mutations. 📌 FPI fellowship, 4 years fully funded!
More information here:
www.bsc.es/join-us/job-...

MMseqs2-GPU sets new standards in single query search speed, allows near instant search of big databases, scales to multiple GPUs and is fast beyond VRAM. It enables ColabFold MSA generation in seconds and sub-second Foldseek search against AFDB50. 1/n
📄 www.nature.com/articles/s41...
💿 mmseqs.com



It was lovely to speak at the CATH 30 symposium, celebrating 30 years of the @cathgene3d.bsky.social protein structure classification database. I was presenting recent work on our new generative protein-family language model: preprint coming soon.
18.09.2025 10:32 — 👍 11 🔁 3 💬 0 📌 0
I also truly appreciated the chance to meet my future colleagues @nbordin.bsky.social, David Miller, Vaishali Waman, @judewells.bsky.social and Ian Sillitoe. I am thrilled to be joining your team soon!
06.08.2025 12:32 — 👍 1 🔁 1 💬 1 📌 0
Poster Prize Awarded at ISMB/ECCB 2025
Congratulations to Jude Wells for Design in voxel space, decode in smiles space: Plixer generates drug-like molecules for protein pockets


Thanks to everyone who came and talked with me about my poster at #PSB2025 : computational methods for predicting which mutations will cause drug inefficacy via protein-drug binding disruption
09.01.2025 01:21 — 👍 5 🔁 0 💬 0 📌 0Does anyone know if Rosetta Interface Analyzer from @rosettacommons.bsky.social is the best method within the Rosetta framework for estimating binding affinity between antibodies and antigens? (Including ddG of mutations)?
14.12.2024 13:31 — 👍 2 🔁 0 💬 0 📌 0
Great blog post from AdaptyvBio summarising submissions for round 2 of the protein design competition: including a few interesting methods I had never heard of. Results will be released tomorrow:
www.adaptyvbio.com/blog/po103
Two BioML starter packs now:
Pack 1: go.bsky.app/2VWBcCd
Pack 2: go.bsky.app/Bw84Hmc
DM if you want to be included (or nominate people who should be!)

A new version of CATH, v4.4, is out! 🎉
Here’s a link to the manuscript in NAR.
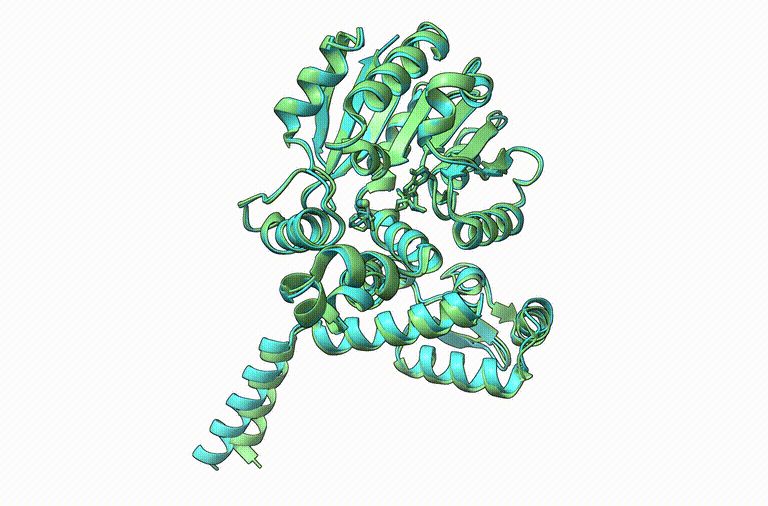
Thrilled to announce Boltz-1, the first open-source and commercially available model to achieve AlphaFold3-level accuracy on biomolecular structure prediction! An exciting collaboration with Jeremy, Saro, and an amazing team at MIT and Genesis Therapeutics. A thread!
17.11.2024 16:20 — 👍 609 🔁 204 💬 18 📌 25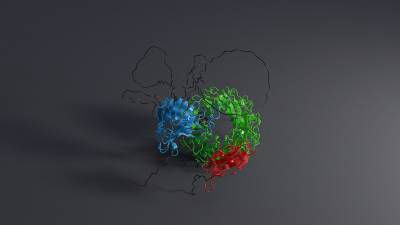
Our recent work TED: The Encyclopedia of Domains showcased by UCL: 365 million domain like structures identified in the AlphaFold DB, 194 million with proposed assignments to CATH superfamilies, plus a catalogue of domain-domain interactions.
www.ucl.ac.uk/computer-sci...








