
Junior, Assistant, or Associate Specialist – Xue Lab
University of California, Irvine is hiring. Apply now!
The Xue lab at UC Irvine is looking for a staff scientist to support our work investigating how microbes interact and evolve in the gut microbiome! Open to a wide range of previous experience levels, see ad for more.
recruit.ap.uci.edu/JPF09601
17.07.2025 20:32 — 👍 116 🔁 112 💬 0 📌 3
Unfortunately we haven’t had the chance to look at other organisms. I think repeat % could matter more if it starts creating large blocks of repetitive sequence (which would then need longer reads). More # of repeat elements should be ok, provided they are surrounded by unique flanking sequence.
21.06.2025 21:05 — 👍 1 🔁 0 💬 0 📌 0

Grateful to be talking at #Evol2025! Will be presenting on how long Nanopore reads need to be in order to accurately call structural variants in Drosophila at the population level. Talk is at 3pm on Saturday in the Genomics III section. If you can’t make it get in touch!
20.06.2025 22:32 — 👍 13 🔁 3 💬 0 📌 0
Many thanks to my co-authors, @hgellert.bsky.social, Jess Smiley-Rhodes, @bernardkim.bsky.social, and @petrovadmitri.bsky.social, without whom this work would not have been possible!
25.04.2025 20:03 — 👍 1 🔁 0 💬 0 📌 0
Our results suggest that reads at least 3x longer than the largest repetitive elements are required to avoid SV-calling errors from these elements. They also highlight that SV-calling is a species-specific problem, as the repeat landscape varies greatly across taxa.
25.04.2025 20:03 — 👍 1 🔁 0 💬 2 📌 0

Finally, we short-read sequenced our inbred lines as the vast majority of genomic data is from NGS. Unsurprisingly, short-read data had the poorest accuracy, as well as the most significant biases against insertions and the largest number of spurious inversion calls.
25.04.2025 20:03 — 👍 0 🔁 0 💬 1 📌 0

We additionally downsampled our 30x-coverage ultra-long reads to 20x- and 10x- coverage. We found that accuracy decreased even at 20x-coverage, and neither low-coverage distribution could recover all three of the cosmopolitan inversions.
25.04.2025 20:03 — 👍 0 🔁 0 💬 1 📌 0
A second significant source of error came from incorrectly merging strain-level SVs into the population call sets. While joint genotyping is well-understood for SNPs, merging SVs is still a highly complex problem that needs to rely on multiple lines of evidence to find that two SVs are the same.
25.04.2025 20:03 — 👍 0 🔁 0 💬 1 📌 0
We were able to determine the cause of false positive SV calls from manual validation. More than half of all errors came from misalignments due to transposable elements (TEs) or complex genomic loci — except for the ultra-long distribution, which had no major issues with TEs or complex regions.
25.04.2025 20:03 — 👍 0 🔁 0 💬 1 📌 0
When we focused on SVs > 10kb, we found that the ultra-long data was very clearly the most accurate. Only the ultra-long data had 100% accuracy when calling large inversions, finding three cosmopolitan inversions in our dataset.
25.04.2025 20:03 — 👍 0 🔁 0 💬 1 📌 0

We report significant shifts in SV-calling accuracy at the population level when systematically varying read length within D. melanogaster. Our ultra-long (as defined by ONT: read N50 > 50kb) read distribution, called more SVs, and at a significantly higher accuracy, than any other distribution.
25.04.2025 20:03 — 👍 0 🔁 0 💬 1 📌 0

As no definitive benchmark SV call sets exist for D. melanogaster, we then manually validated more 2,300 SVs at over 18,000 genomic loci across the read-length distributions to assess variant-calling accuracy. Validation was done by visualizing read alignments in Jbrowse2.
25.04.2025 20:03 — 👍 0 🔁 0 💬 1 📌 0
Using a combination of read-based (Sniffles2, cuteSV, DeBreak) and assembly-based callers (svim-asm, PAV), we called structural variants in every line and read-length distribution. Each strain's calls were merged together (Jasmine) for each distribution, creating a pop. level variant call set.
25.04.2025 20:03 — 👍 0 🔁 0 💬 1 📌 0
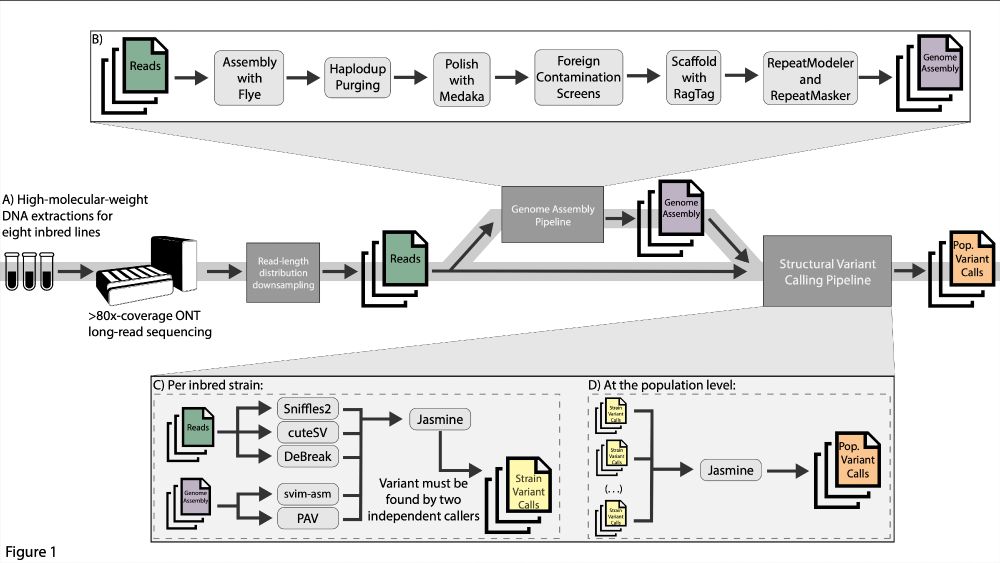
To investigate this, we Nanopore sequenced eight D. melanogaster inbred lines to extremely high coverage (mean 238x) and then downsampled the reads to create 30x-coverage pools of distinct read-length distributions (as quantified by read N50). We additionally assembled genomes for each pool.
25.04.2025 20:03 — 👍 0 🔁 0 💬 1 📌 0
Just as a drastic impact on structural variant calling was seen moving from short reads to long reads, it is likely that the accuracy of structural variant calling significantly varies along the spectrum of read lengths produced by long-read sequencing methods.
25.04.2025 20:03 — 👍 0 🔁 0 💬 1 📌 0
@nanoporetech.com long-reads can range 1000-fold in length (100s bp - 1Mb) in a single sequencing run, but the consequences of significantly different long-read lengths on the accuracy of genome-wide structural variant calling is not well understood, especially for non-human species.
25.04.2025 20:03 — 👍 0 🔁 0 💬 1 📌 0
The increasing availability of long-read data is leading to a boom in population-level SV datasets. These datasets are critical for uncovering SV polymorphisms, which better capture the dynamic evolutionary processes that shape SV diversity.
25.04.2025 20:03 — 👍 0 🔁 0 💬 1 📌 0
Thrilled to see this work, led by @lisacouper.bsky.social now out!
We quantified variation in thermal tolerance in the mosquito, Aedes sierrensis, to quantify how adaptation may alter disease vector distributions under warming. 🧵
www.pnas.org/doi/10.1073/...
21.01.2025 15:56 — 👍 18 🔁 7 💬 1 📌 0
PhD candidate in Dmitri Petrov's lab @ Stanford
genomics @ stanford
gpreising.github.io
he/him
Evolutionary systems/cell biologist. EMBO and SNSF Postdoctoral Fellow with Dmitri Petrov and Dan Jarosz at Stanford. PhD with Andreas Wagner at the University of Zurich. Studying how molecular and cellular systems shape, and are shaped by, evolution.
Evolutionary geneticist
Adaptation to toxins across environments and timescales
K99 Fellow| Marie Curie Fellow
https://www.mariannakarageorgi.com/
I am an Assistant Professor of Biology at the University of Vermont. My lab works on fundamental questions of population genetics as well as ecological and evolutionary genomics.
https://www.jcbnunez.org/
Stanford Law prof, working on ethical, legal, and social issues in the biosciences. Sports fan (especially Stanford sports). And strongly opposed to the illegal & immoral Russian invasion of Ukraine. Slava Ukraini!
Focusing the power of genomics to create a healthier world. Leader in pangenomics, conservation genomics, cancer genomics, pathogen genomics, and nanopore sequencing. Home of the UCSC Genome Browser, @ucscxena, UShER, Dockstore, and other high-power tools.
Associate Professor of Genetics and Genome Sciences at UConn Health and The Jackson Laboratory
Transposons, Structural Variation, Genomics, Gardening, Loud Music, Mom
Postdoctoral Researcher
Kim Lab @ Princeton EEB
Evolutionary Biology. Adaptation Genomics. Transposable Elements. Drosophila. @IBB_Botanic @CSIC @eseb_org Vice President @Dros_EU @LCATMon #melanogasterCTF #ERCgrantee
Lab web: gonzalezlab.eu
Science outreach: www.melanogaster.eu
DrosEU: www.droseu.net
Lecturer and DECRA Fellow at the University of Sydney
Invasion genomics || Wolbachia || Dispersal
https://scholar.google.com.au/citations?user=ipvZJRQAAAAJ&hl
The UCSC Genome Browser is a public, freely available, open-source web-based graphical viewer for displaying genome sequences and their annotations.
lover of birds, nature, evolutionary biology, diversity in STEM, peace and good will
Post-doc @HarvardOEB – Evolutionary Genomics | PhD @helsinkiuni
PhD student in Fordyce & Petrov labs at Stanford | biophysics of evolutionary adaptation in proteins | using population genomics, synthetic biology, microfluidics
Dad, bioinformatician, ex-expat. Currently working on transposon bioinformatics & infectious disease genomics.
Postdoc at @UBuffalo | Gokcumen Lab
Evolution, structural variants and population genetics.
PhD from @UABBarcelona | Inversion polymorphism.
https://biolevol.github.io/
evolutionary biologist at NYU Biology, lab website: https://shchurch.github.io/
Evolutionary geneticist. I investigate genome evolution, sex chromosomes, and spermatogenesis across mammals and fish. https://www.danshawgenes.com/
Menswear writer. Editor at Put This On. Words at The New York Times, The Washington Post, The Financial Times, Esquire, and Mr. Porter.
If you have a style question, search:
https://dieworkwear.com/ | https://putthison.com/start-here/























