AlphaFold is amazing but gives you static structures 🧊
In a fantastic teamwork, @mcagiada.bsky.social and @emilthomasen.bsky.social developed AF2χ to generate conformational ensembles representing side-chain dynamics using AF2 💃
Code: github.com/KULL-Centre/...
Colab: github.com/matteo-cagia...
17.04.2025 19:10 — 👍 205 🔁 63 💬 3 📌 4
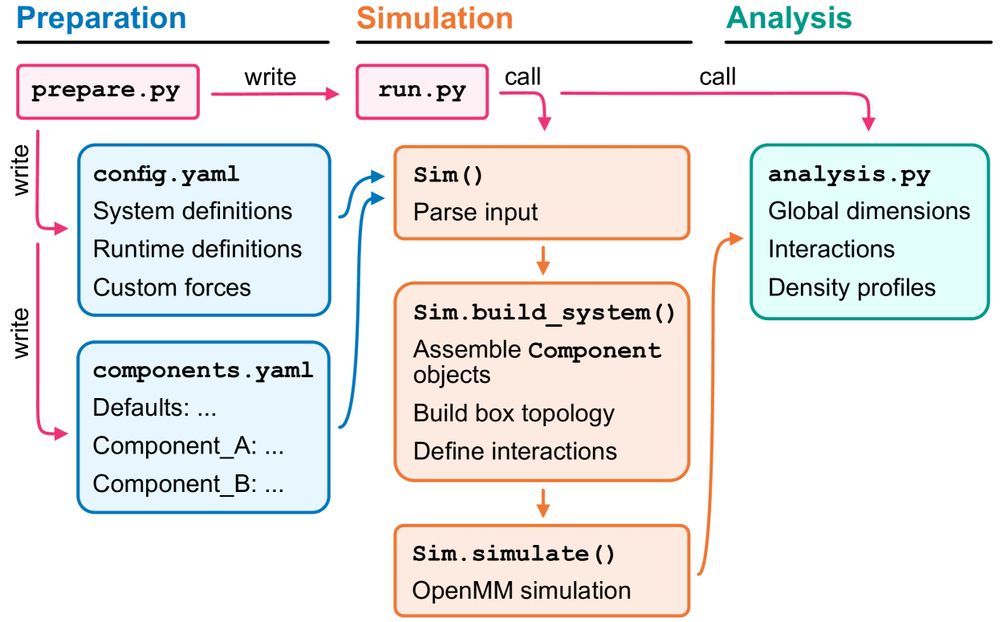
Figure showing the architecture of the CALVADOS package.
Do you like CALVADOS but are not quite sure how to make it?
We’ve got your back!
@sobuelow.bsky.social & @giuliotesei.bsky.social—together with the rest of the team—describe our software for simulations using the CALVADOS models incl. recipes for several applications. 1/5
doi.org/10.48550/arX...
15.04.2025 07:08 — 👍 47 🔁 16 💬 2 📌 2
📢 Our article calling for a #FAIR database for #MolecularDynamics simulation data has now been peer-reviewed and published in @naturemethods.bsky.social
📖 Read it here: rdcu.be/ef6YX
📝 Support the statement: bit.ly/3zVS3qm
#MDDB #FAIRdata #collaboration
04.04.2025 08:09 — 👍 37 🔁 21 💬 0 📌 3

HIV Protein Switch May Help Virus Squeeze into Host Cell Nucleus
Simulations on Bridges-2 Help Pitt Team Visualize Rare, Transient Shape Change in Capsid Protein
And huge thanks to Ken Chiacchia and Jorge Salazar for highlighting our work! Check out their articles for a breakdown of the paper :)
www.psc.edu/hiv-1-capsid...
tacc.utexas.edu/news/latest-...
27.03.2025 17:50 — 👍 3 🔁 1 💬 0 📌 0

The bulk of my thesis work was recently published!
We used 19F NMR and weighted ensemble simulations among other methods to explore hidden dimer states of the HIV-1 capsid protein.
If this sounds interesting to you, see the full paper here:
www.pnas.org/doi/10.1073/...
27.03.2025 17:50 — 👍 11 🔁 1 💬 1 📌 0
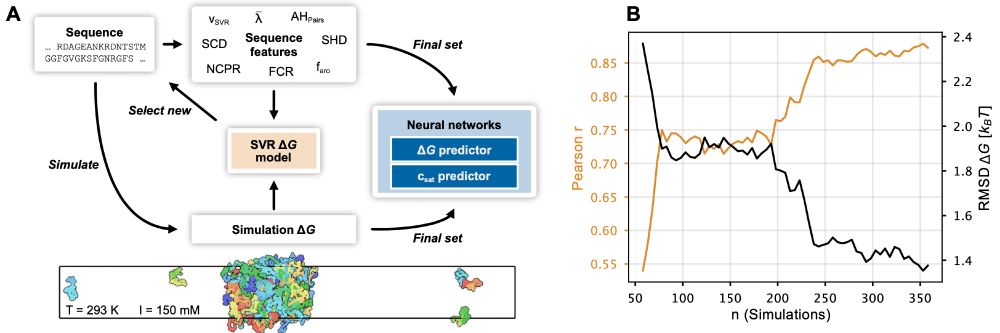
Our paper on prediction of phase-separation propensities of disordered proteins from sequence is now published:
www.pnas.org/doi/10.1073/...
The paper has been substantially updated compared to the preprint including new experimental data and using the neural network to finetune CALVADOS. 1/n
25.03.2025 17:55 — 👍 71 🔁 17 💬 1 📌 1
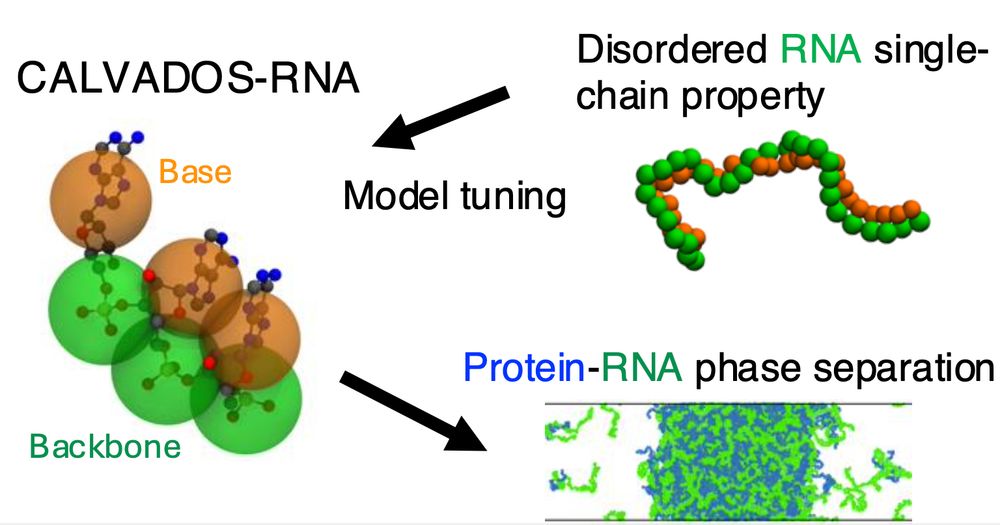
Table of Contents figure showing the CALVADOS-RNA model and a snapshot from a mixed protein-RNA condensate
CALVADOS-RNA is now published
doi.org/10.1021/acs....
This is a simple model for flexible RNA that complements and works with the CALVADOS protein model. Work led by Ikki Yasuda who visited us from Keio University.
Try it yourself using our latest code for CALVADOS
github.com/KULL-Centre/...
26.02.2025 19:11 — 👍 67 🔁 20 💬 1 📌 0
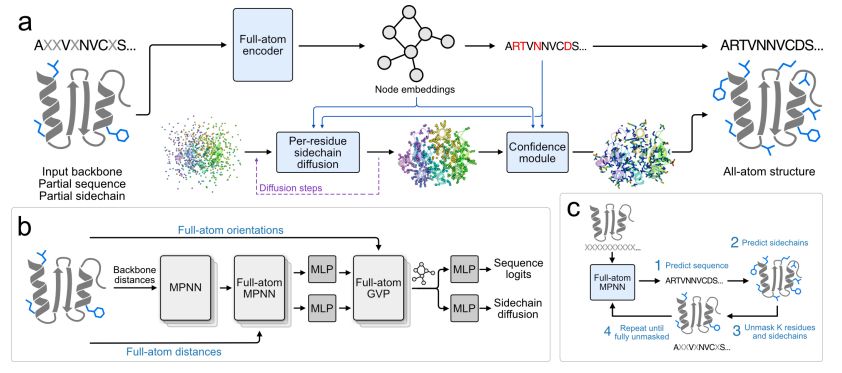
FAMPNN architecture
All-atom fixed backbone protein sequence design with FAMPNN
@richardshuai.bsky.social Talal Widatalla @possuhuanglab.bsky.social @brianhie.bsky.social
www.biorxiv.org/content/10.1...
21.02.2025 22:37 — 👍 30 🔁 7 💬 0 📌 0
The BioEmu-1 model and inference code are now public under MIT license!!!
Please go ahead, play with it and let us know if there are issues.
github.com/microsoft/bi...
19.02.2025 20:17 — 👍 103 🔁 39 💬 2 📌 2
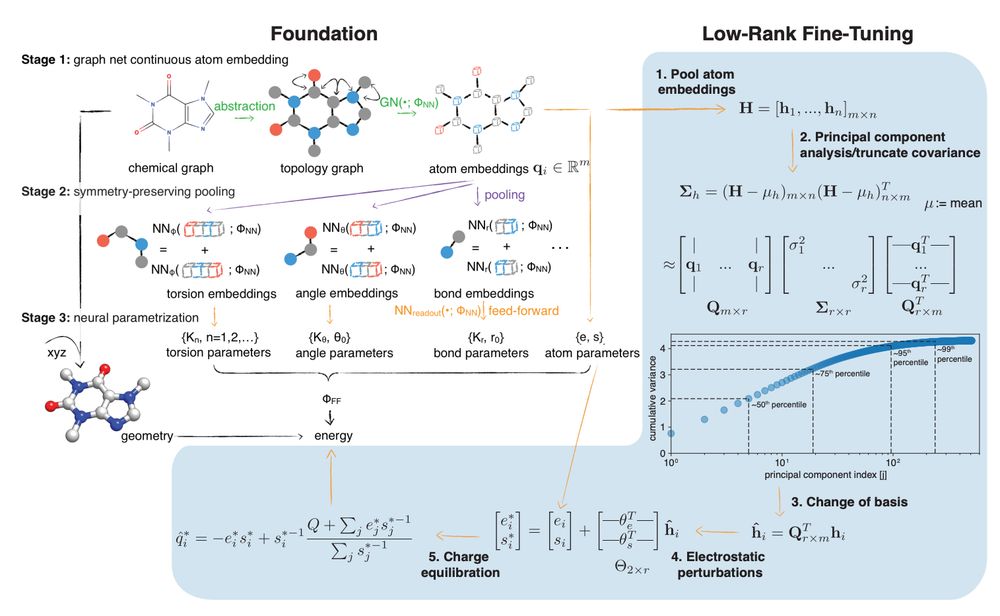
Figure 1 from arXiv preprint https://doi.org/10.1101/2025.01.06.631610
Fig. 1 Espaloma is an end-to-end differentiable molecular mechanics parameter assignment scheme for arbitrary organic molecules. Espaloma (extensible surrogate potential optimized by message-passing) is a modular approach for directly computing molecular mechanics force field parameters FFF from a chemical graph G such as a small molecule or biopolymer via a process that is fully differentiable in the model parameters FNN. In Stage 1, a graph neural network is used to generate continuous latent atom embeddings describing local chemical environments from the chemical graph. In Stage 2, these atom embeddings are transformed into feature vectors that preserve appropriate symmetries for atom, bond, angle, and proper/improper torsion inference via Janossy pooling.54 In Stage 3, molecular mechanics parameters are directly predicted from these feature vectors using feed-forward neural networks. This parameter assignment process is performed once per molecular species, allowing the potential energy to be rapidly computed using standard molecular mechanics or molecular dynamics frameworks thereafter. The collection of parameters FNN describing the espaloma model can be considered as the equivalent complete specification of a traditional molecular mechanics force field such as GAFF38,39/AM1-BCC55,56 in that it encodes the equivalent of traditional typing rules, parameter assignment tables, and even partial charge models. Reproduced from ref. 49 with permission from the Royal Society of Chemistry.
Everything is chaos, but I wanted to share some awesome recent science from the lab that hints at where the future of biomolecular simulation is headed:
Foundation simulation models that can be fine-tuned to experimental free energy data to produce systematically more accurate predictions.
19.02.2025 19:30 — 👍 107 🔁 30 💬 3 📌 1
New paper from our lab @naturecomms.bsky.social!
We reveal the dynamics and mechanism of target DNA traversal in #CRISPR Cas12a, a conundrum in the field!
nature.com/articles/s41...
#compchem
We thank the amazing #HPC resources of PSC #Anton2 and SDSC
08.02.2025 18:22 — 👍 46 🔁 9 💬 3 📌 0
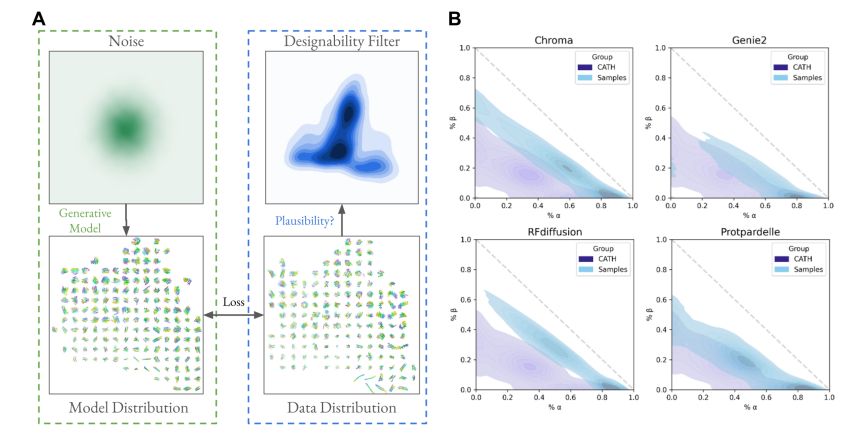
Generative models capture a biased set of protein structure space
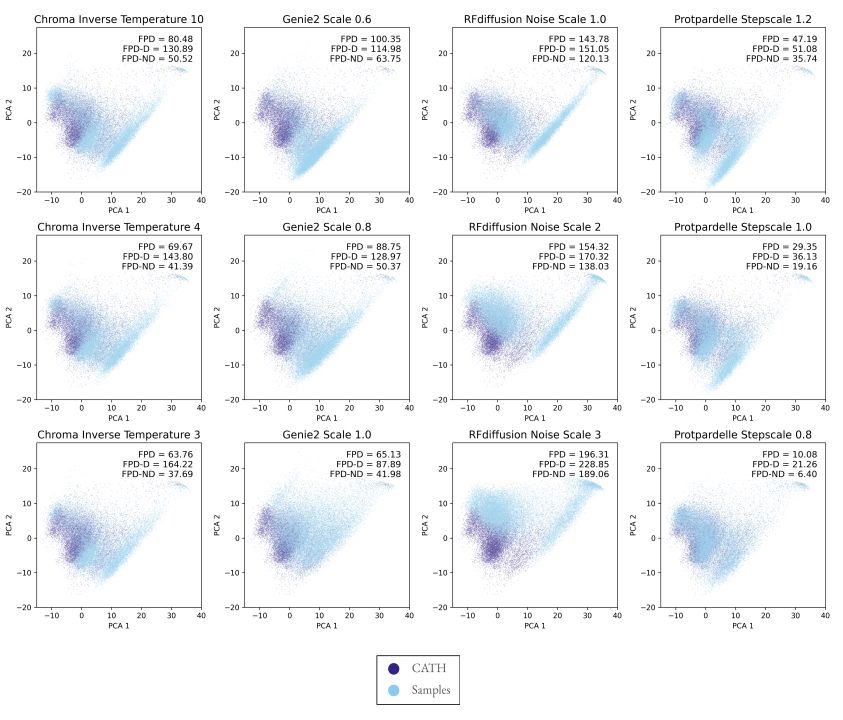
Generative models do not capture the full expressivity of PDB structures
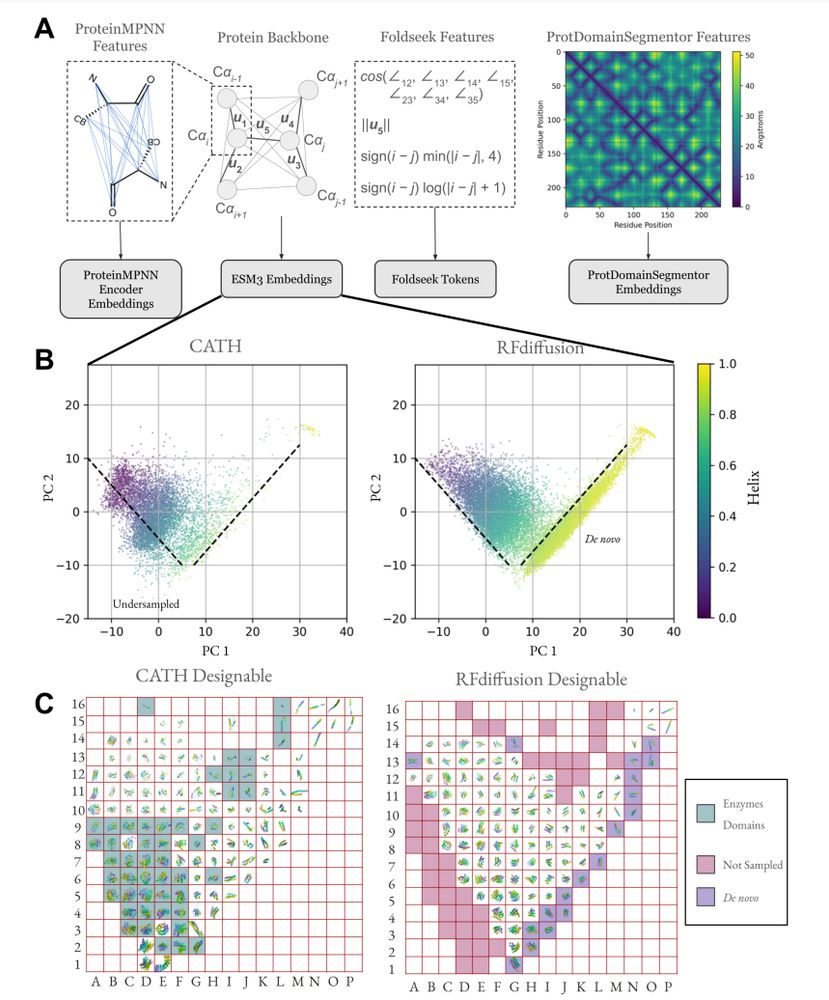
Protein structure embeddings reveal undersampled and de novo structure space
A framework for evaluating how well generative models of protein structure match the distribution of natural structures.
@possuhuanglab.bsky.social
www.biorxiv.org/content/10.1...
15.01.2025 23:10 — 👍 43 🔁 10 💬 0 📌 0

Figure from the paper that illustrates the approach of probing the transition state for amyloid growth by experiments and simulations
How do proteins mis-fold?
Paper led by Jacob Aunstrup from Alex Büll’s lab with MD simulations by Abigail Barclay, and key contributions from several others. We combined measurements of Φ-values with MD simulations to study the transition state for amyloid fibril growth
doi.org/10.1038/s415...
16.01.2025 15:09 — 👍 91 🔁 22 💬 1 📌 2


🚨 Revolutionising Snakebite Treatments with AI-Designed Proteins 🐍
I'm proud to share our latest study published in hashtag#Nature, driven by Susana Vazquez Torres, and co-led by David Baker (Institute for Protein Design, University of Washington) and myself.
15.01.2025 20:16 — 👍 27 🔁 11 💬 2 📌 2
move over ligand RMSD < 2 Å 😤 ConfBench is on the scene!
if you're interested in the evaluation of conformational accuracy of structure prediction methods, take a look at our first stab at a systematic conformational benchmark in the NP3 technical report below! 🧵
www.iambic.ai/post/np3-tec...
17.12.2024 04:37 — 👍 49 🔁 15 💬 1 📌 1
Professor @pittchem.bsky.social. Computational biophysicist, leading @westpasoftware.bsky.social development for weighted ensemble rare-event sampling, Amber force field developer.
https://westpa.github.io
Open-source, highly scalable software for running weighted ensemble simulations with any dynamics engine, including MD (e.g., Amber, OpenMM) and systems biology engines (BioNetGen). We are part of the @omsf.bsky.social consortium.
Working with data in the molecular sciences. https://www.mdanalysis.org
Independent news from the University of Copenhagen and beyond. Got a story? Mail to uni-avis@adm.ku.dk.
Advanced molecule editor and visualizer
Currently working towards Avogadro2
https://two.avogadro.cc/
https://discuss.avogadro.cc/
News and information from the European Commission. Social media and data protection policy: http://europa.eu/!MnfFmT
Protein scientist in Copenhagen
Associate Professor of Machine Learning and Signal Processing, Technical University of Denmark (DTU)
https://frellsen.org
Founder & CEO @jura.bsky.social | Full-stack probabilistic machine learning for the development of genetic medicines | NYC & Basel & Boston
Full-stack machine learning and synthetic biology company dedicated to transforming personalized, immune-mediated medicine
Assistant professor of chemistry at the Technical University of Denmark (DTU). Also at Jura Bio. machine learning, statistics, chemistry, biophysics
https://eweinstein.github.io/
The Department of Chemistry in the Dietrich School of Arts and Sciences at the University of Pittsburgh. A leader in chemical research and education since 1875
Computational Structural Biologist
Assistant Professor @VanderbiltMPB
wankowiczlab.com
(she/her)
Past: UCSF, Dana-Farber, Broad Institute, UMass Amherst
America’s Finest News Source. A @globaltetrahedron.bsky.social subsidiary.
Get the paper delivered to your door: membership.theonion.com
Join The Onion Newsletter: https://theonion.com/newsletters/
Illuminating math and science. Supported by the Simons Foundation. 2022 Pulitzer Prize in Explanatory Reporting. www.quantamagazine.org
Husband, Father, Grandfather, Datahound, Dog lover, Fan of Celtic music, Former NIGMS director, Former EiC of Science, Stand Up for Science advisor, Shenanigator, Pittsburgh, PA
NIH Dashboard: https://jeremymberg.github.io/jeremyberg.github.io/index.html
Research group led by Charlotte Deane, based in the Department of Statistics at the University of Oxford.
https://opig.stats.ox.ac.uk/
Molecular Dynamics Data Bank. The European Repository for Biosimulation Data.
Funded by the European Union
www.mddbr.eu