The workshop "Quantum Chemistry for Drug Design: From Theory to Applications", which we organized at IOCB Prague, has just concluded. Leading academics and pharmaceutical industry practitioners came together to share their knowledge and insights.
Thanks to everybody who made it happen!
12.09.2025 10:13 — 👍 1 🔁 0 💬 0 📌 0

Day 3 opened with Kenneth Atz @Roche tackling the holy grail of #CADD: P-L binding #affinity prediction. By reframing limits of data & models, we can focus on the next solvable challenges - a sharp reminder of complexity & progress ahead.
#CECAM @cecamevents.bsky.social @iocbprague.bsky.social
10.09.2025 09:31 — 👍 6 🔁 2 💬 0 📌 0

We opened Day 2 of our #CECAM flagship workshop in Prague with the CECAM director Andrea Cavalli, highlighting steered #MD, dynamical docking & the complexity of binding energetics, and the challenges ahead. 🚀
#CECAMinPrague @iocbprague.bsky.social @cecamevents.bsky.social @iocbtech.bsky.social
09.09.2025 11:20 — 👍 9 🔁 4 💬 0 📌 0

PM6-ML, our semiempirical quantum-mechanical #CompChem method with machine learning correction (see paper: pubs.acs.org/doi/10.1021/...), is now also available as an Atomic Simulation Environment (ASE) calculator.
github.com/Honza-R/PM6-...
15.07.2025 11:16 — 👍 18 🔁 3 💬 0 📌 0

I added the g-xTB #compchem method, just introduced by @grimmelab.bsky.social, to our protein-ligand interaction energy benchmarking. With an average error of less than 5% in the PLA 15 dataset, it is the most accurate semiempirical QM method to date (when ML is not considered).
07.07.2025 12:51 — 👍 10 🔁 0 💬 1 📌 0
I didn't know you were into this! We'll have something to talk about next time we meet.
04.07.2025 11:21 — 👍 1 🔁 0 💬 0 📌 0
I agree that the LLM has no intent (fingers crossed it doesn't). However, the training data probably contains plenty of "unsupported excuses" to draw from. So, to be precise, it's not lying. It's merely reproducing previously existing lies.
02.07.2025 18:48 — 👍 1 🔁 0 💬 0 📌 0
2/2 This was clearly a deliberate lie. Other queries on the same API worked, and when I ran the code, the connection was fine, but there were other, quite obvious, errors. It was a reasoning model that can run the code it works on, so it likely saw the same output as me but tried to find an excuse.
02.07.2025 09:39 — 👍 1 🔁 0 💬 1 📌 0
1/2 Lying is not the same as hallucinating. I asked an LLM to write a script to fetch data from a public API. After a couple of iterations, during which I fixed the issues and the AI apologized, it started telling me that the code was correct, but that it was having trouble connecting to the API.
02.07.2025 09:39 — 👍 1 🔁 0 💬 1 📌 0

Benchmark of Approximate Quantum Chemical and Machine Learning Potentials for Biochemical Proton Transfer Reactions
Proton transfer reactions are among the most common chemical transformations and are central to enzymatic catalysis and bioenergetic processes. Their mechanisms are often investigated using DFT or approximate quantum chemical methods, whose accuracy directly impacts the reliability of the simulations. Here, a comprehensive set of semiempirical molecular orbital and tight-binding DFT approaches, along with recently developed machine learning (ML) potentials, are benchmarked against high-level MP2 reference data for a curated set of proton transfer reactions representative of biochemical systems. Relative energies, geometries, and dipole moments are evaluated for isolated reactions. Microsolvated reactions are also simulated using a hybrid QM/MM partition. Traditional DFT methods offer high accuracy in general but show markedly larger deviations for proton transfers involving nitrogen-containing groups. Among approximate models, RM1, PM6, PM7, DFTB2-NH, DFTB3, and GFN2-xTB show reasonable accuracy across properties, though their performance varies by chemical group. The ML-corrected (Δ-learning) model PM6-ML improves accuracy for all properties and chemical groups and transfers well to QM/MM simulations. Conversely, standalone ML potentials perform poorly for most reactions. These results provide a basis for evaluating approximate methods and selecting potentials for proton transfer simulations in complex environments.
🚀 Benchmark paper out!
How well do DFT, semiempirical & ML methods model proton transfer?
✅ DFT performs well, except with N-groups
❌ Pure ML struggles (though ORB v3 shows big gains)
🔥 PM6-ML Δ-learning excels, even in QM/MM setups!
Check it out: pubs.acs.org/doi/10.1021/...
01.07.2025 12:37 — 👍 6 🔁 1 💬 0 📌 0
I'm at the WATOC #CompChem conference in Oslo. Machine learning is everywhere, but the hottest news so far is the new g-xTB method by @grimmelab.bsky.social . The results presented today are truly impressive. I'm already running first calculations on our biomolecular systems...
26.06.2025 09:39 — 👍 18 🔁 3 💬 0 📌 1

GitHub - pdynamo/pDynamo3: The pDynamo molecular modeling and simulation program
The pDynamo molecular modeling and simulation program - pdynamo/pDynamo3
PM6-ML, our latest method that aims for quantum-chemical accuracy in large biomolecular systems, has a third implementation. In addition to MOPAC-ML and Cuby4, PM6-ML is now available in pDynamo3, where it can be used for QM/MM calculations: github.com/pdynamo/pDyn....
19.05.2025 06:47 — 👍 11 🔁 1 💬 1 📌 0
A humble #compchem contribution to a great experimental #medchem work ranging from novel synthesis protocol to in vivo models. We applied our SQM-based scoring to interpret the interaction of the novel inhibitors with the protein.
12.05.2025 11:15 — 👍 6 🔁 0 💬 0 📌 0
Our PM6-ML method, a semiempirical QM method with ML correction, works well for proton transfer reactions - despite not having been trained for that. The new implementation reported in the preprint allows its use in QM/MM biomolecular simulations.
05.05.2025 12:08 — 👍 4 🔁 1 💬 0 📌 0
CECAM - Quantum Chemistry for Drug Design: From Theory to Applications
🚀 Exciting news! We're organizing a @cecamevents.bsky.social Flagship Workshop on Quantum Chemistry in Drug Design in Prague, Sept 8–10, 2025!
Join top experts from academia & industry. Few spots left for contributed talks!
📢 Apply now: www.cecam.org/workshop-det...
#compchem #cadd #QM #CECAM
24.03.2025 12:16 — 👍 12 🔁 6 💬 0 📌 0
A perspective on the importance (and the lack of) reliable benchmarks for structure-based computer-aided drug design methods - with a contribution of @adampecina.bsky.social from my group
10.03.2025 12:37 — 👍 2 🔁 0 💬 0 📌 0
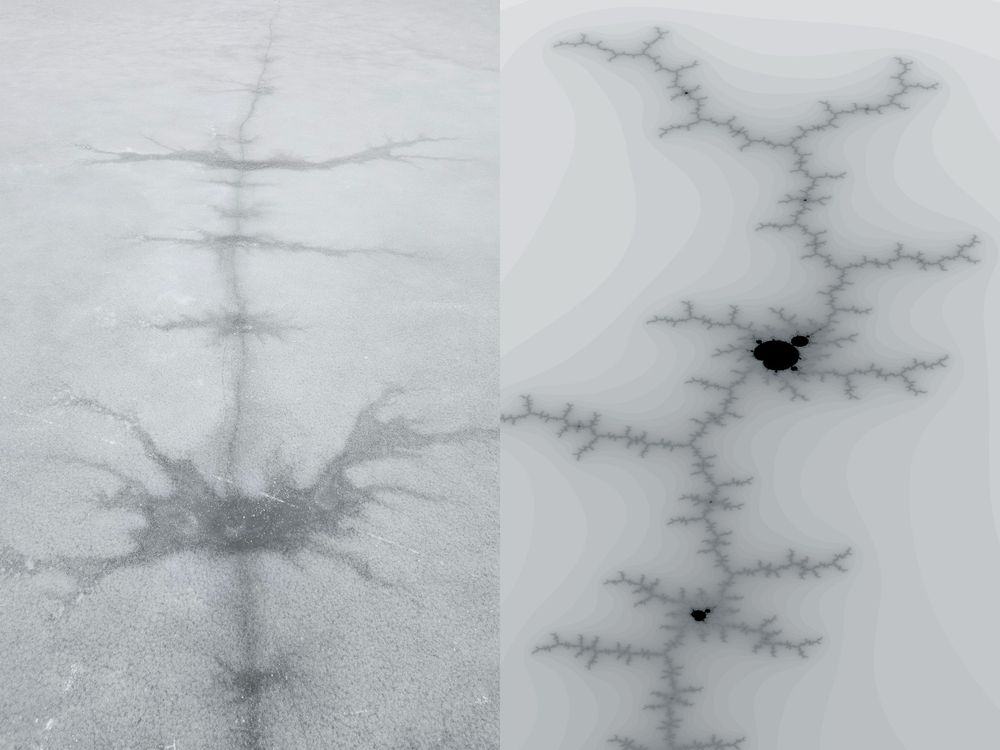
patterns in ice similar to a fractal rendered next to it
Connecting #skating and #science - a photo of the ice we skated on Sunday (left) and the Mandelbrot set fractal (right)
21.01.2025 12:44 — 👍 3 🔁 0 💬 0 📌 0
We're organizing a CECAM workshop in September. If you're interested in QM calculations for drug design, apply and join us in Prague:
www.cecam.org/workshop-det...
15.01.2025 11:49 — 👍 4 🔁 2 💬 0 📌 0
OpenBind is building the world’s largest open-access dataset of drug–protein interactions to speed up new treatments. Through cutting-edge science and open collaboration, we power AI tools for structure-based drug design – free for all.
🧬 Structural Bioinformatics | 💊 AI/ML for Drug Discovery | Geometric DL
🔬 @iocbprague.bsky.social, prev. PhD @cusbg.bsky.social @mff.unikarlova.cuni.cz
Professor & Scientist @uspoficial.bsky.social 🇧🇷. Visiting @mit.edu. Scholar @ieausp.bsky.social. Computational (bio)chemistry and biophysics.
Web: http://gaznevada.iq.usp.br
🐦 : x.com/ArantesLab
🎵 : www.dinamicas.art.br
Associate Professor Sci Informatics & Biochemistry at Virginia Tech. Comp. Chem, CADD, & Applied Data Science. Opinions my own.
World-class science with Czech roots.
linktr.ee/czexpats
The Czech Academy of Sciences (AV ČR) is a complex of 54 public research institutions in the Czech Republic that engage in basic and applied research. | www.avcr.cz/en
#biology #chemistry #geology #geography #enviromentalsciences
Prague, Czechia
web: https://natur.cuni.cz/en
We study nanomaterials experimentally and biological systems using computational chemistry
https://www.kfc.upol.cz/en/
Our online lectures:
https://youtube.com/@katedrafyzikalnichemie7978?si=NCXez1EcHYHXV083
playmolecule.org - Molecular discovery for everyone.
physical and computational chemist with interest in photochemistry, radiation chemistry and X-ray technologies
Computational chemist at the University of Copenhagen #compchem
FACCTs is bringing the ORCA quantum chemistry software to industry - promoting the next Quantum leaps in the Pharma, Materials and Chemical Industries.
Visit us at www.faccts.de
(ML ∪ Bayesian optimization ∪ active learning) ∩ (drug discovery)
Researcher @valenceai.bsky.social
Details: austintripp.ca
Computational Biophysicist, Amateur Photographer, History+Language Nerd.
All views my own.
Website: https://martinvoegele.github.io/
Achira | http://achira.ai
Research laboratory | http://choderalab.org
Antiviral drug discovery for pandemics | http://asapdiscovery.org
OpenADMET | http://openadmet.org
Employer-mandated disclaimer: http://choderalab.org/disclaimer
Pronouns: he/him
Computational chemistry professor at @unistuttgart.bsky.social simulating reaction mechanisms, atom tunneling, astrochemistry, MachineLearning.
https://www.uni-stuttgart.de/theochem/kaestner
Science journalist & writer, https://denikn.cz, Prague, Czechia
Group Leader at the Institute of Organic Chemistry and Biochemistry (IOCB Prague @iocbprague.bsky.social). Plant biochemistry, metabolomics, genomics, bioML, etc. https://www.pluskal-lab.org
Proud dad, structural bioinformatician, cook, birdwatcher and skater