
Thanks so much to @bribarbu.bsky.social and @cenmag.bsky.social for highlighting our recent work! 😊
@tkschramm.bsky.social @unibonn.bsky.social
Thanks so much to @bribarbu.bsky.social and @cenmag.bsky.social for highlighting our recent work! 😊
@tkschramm.bsky.social @unibonn.bsky.social
Congrats to everyone involved!
10.09.2025 12:58 — 👍 3 🔁 0 💬 1 📌 0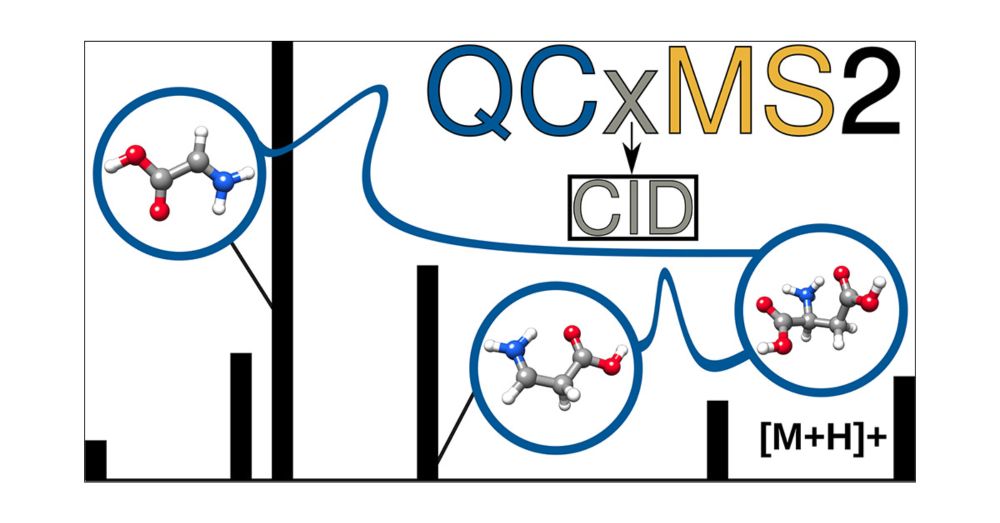
QCxMS2 can now also simulate CID mass spectra.
Just published in #JASMS : doi.org/10.1021/jasms.5c00234
Grateful to my coauthors Stefan Grimme @grimmelab.bsky.social & Marianne Engeser @unibonn.bsky.social - this is the last project of my PhD and completes my work on QCxMS2!
#MassSpec #compchem
🐍 𝗠𝗶𝗻𝗱𝗹𝗲𝘀𝘀𝗚𝗲𝗻 (GitHub): github.com/grimme-lab/M...
Many thanks to everyone involved!

🚀 JCIM: "Chemical Space Exploration with Artificial Mindless Molecules"
We present MindlessGen, an open-source tool for generating chemically diverse "mindless" molecules, and the MB2061 benchmark set with high-level reference data to test methods on unconventional systems.
doi.org/10.1021/acs....
Congrats André and all the authors involved in this new generation of SBDIPY and BIDIPY and their application in photocatalysis.
t.co/0B4FRFBAUy
You can now use g-xTB @grimmelab.bsky.social with ORCA via the ExtOpt feature! Check out our new tutorial and learn how to use it in GOAT, NEB-TS and more.
www.faccts.de/docs/orca/6....
#ORCAqc #FACCTs #gxTB #CompChem #QuantumChem
I'm at the WATOC #CompChem conference in Oslo. Machine learning is everywhere, but the hottest news so far is the new g-xTB method by @grimmelab.bsky.social . The results presented today are truly impressive. I'm already running first calculations on our biomolecular systems...
26.06.2025 09:39 — 👍 18 🔁 3 💬 0 📌 1
After years of development and preparatory works which you might have seen on this profile, a major milestone is achieved:
g-xTB marks not just an evolution, but a revolution in the capabilities of semiempirical quantum chemistry. Convince yourself! A thread.
🔗 chemrxiv.org/engage/chemr...
#compchem

After almost 3 years of development with @grimmelab.bsky.social, a first preliminary version of our next-generation general extended Tight-Binding (g-xTB) is now on ChemRxiv!
Catch the details at #WATOC: my talk (Thu Session B1) and Stefan’s talk (Thu Session A2).
#compchem
doi.org/10.26434/che...
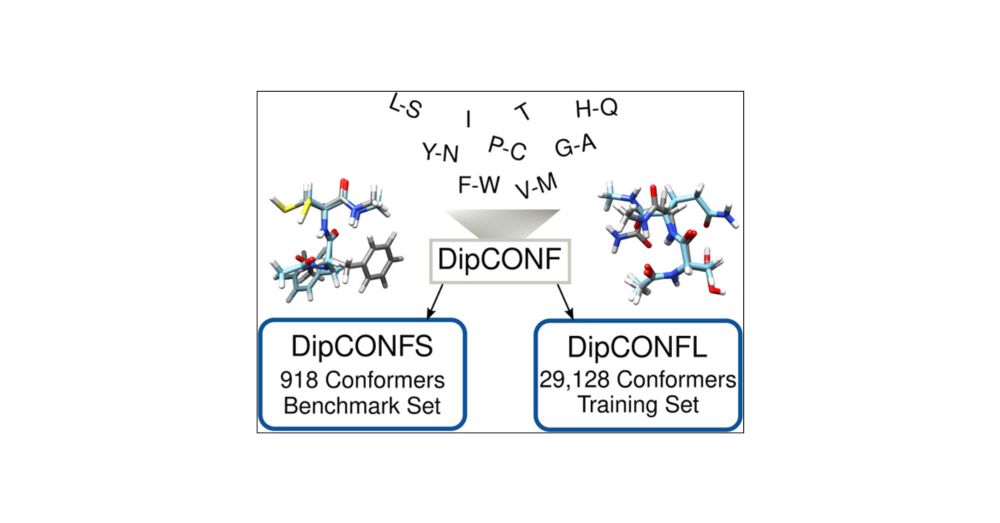
Have you ever wondered how accurate reference data for training ML models is?
Check out the DipCONFS, a set of highly accurate conformational energies for nearly 1,000 dipeptide conformers, and the DipCONFL with ~30,000 high-quality DFT data points.
pubs.acs.org/doi/full/10....
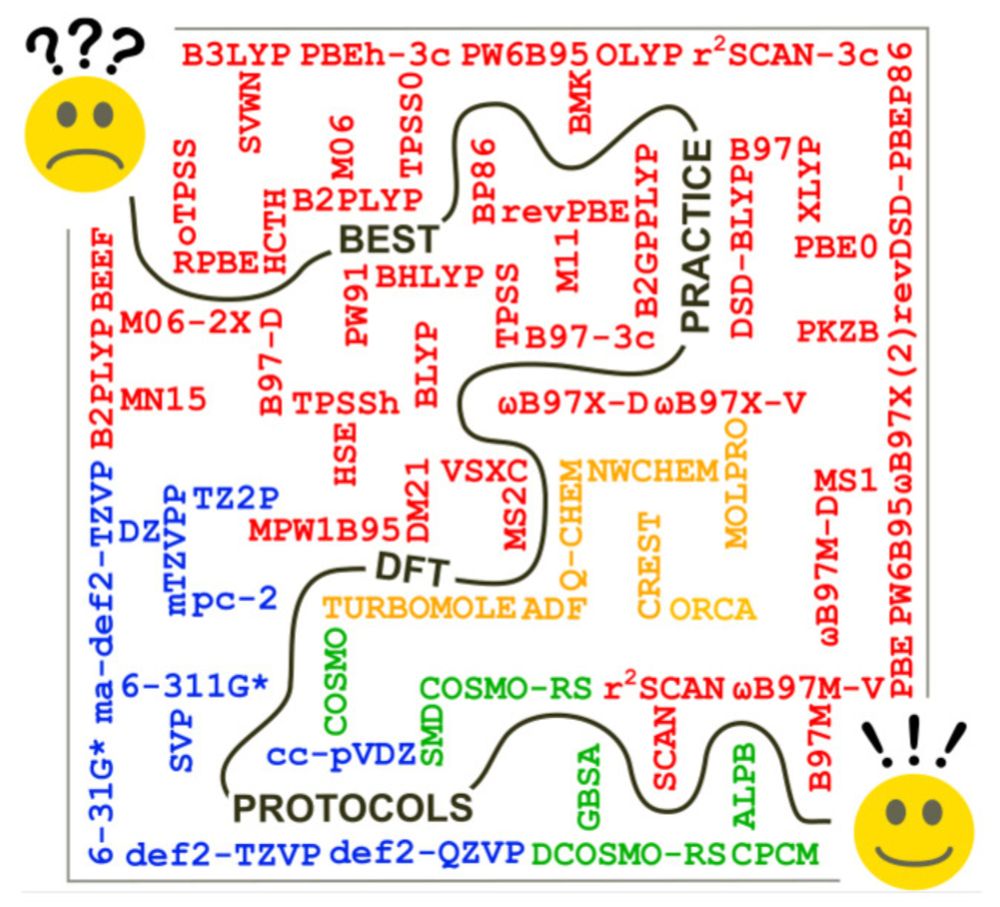
I realized that a #compchem DFT best-practice article was lacking in the literature, so I invited @grimmelab.bsky.social @dermewes.bsky.social @markusbursch.bsky.social and co-workers to write the article below. It was and remains super popular and well received. Great work! (9/22)
05.04.2025 10:30 — 👍 25 🔁 5 💬 2 📌 1
Congratulations @grimmelab.bsky.social for an impressive 6th place across all disciplines: topresearcherslist.com
With #compchem (DFT-D) taking first place in chemistry, and Georg Kresse (VASP/PAW-PPs/DFT) taking the 2nd place (overall, 1st in physics), I'd say:
DFT is winning science :)
Check out our pre-print on accelerated semi-empirical electronic structure theory calculations on consumer-grade GPUs! ⬇️
07.03.2025 10:04 — 👍 6 🔁 2 💬 0 📌 0
Check out our new EEQBC model!
It delivers accurate and robust atomic charges for all elements up to Z=103. By incorporating bond capacitors, we eliminate most artificial CT while preserving the simplicity and efficiency of classical charge equilibration:
doi.org/10.26434/che...
#compchem
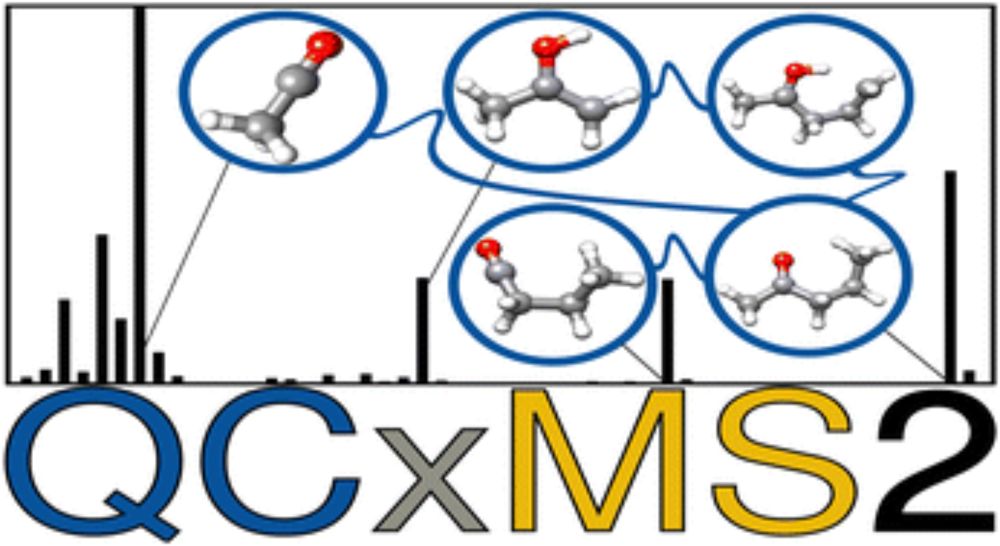
Our paper @grimmelab.bsky.social on QCxMS2 for the calculation of EI-MS was just accepted by @pccp.rsc.org Check out the final article: pubs.rsc.org/en/content/a...
#CompChemSky #MassSpecSky
A unique opportunity to collaborate with our group, the Grimme lab, and Prof. Frank Neese’s department at MPI-KOFO!
Join us in integrating our latest semiempirical method, g-xTB, into ORCA – unlocking access to even more molecular properties. 📈
Interested? Apply now! 📧
Please reshare this post 🚀

The quantum chemical prediction of EI-MS spectra is an important challenge. Check out @grimmelab.bsky.social's new preprint on their QCxMS2 program using ORCA's QM portfolio and NEB-TS infrastructure.
doi.org/10.26434/che...
#ORCAqc #ChemSky #CompChemSky #MassSpectrometry
QCxMS2, the successor to QCxMS, is now available for calculating electron ionization mass spectra using quantum mechanical methods!
05.02.2025 13:59 — 👍 16 🔁 6 💬 2 📌 0
So that's it! We wrapped up the 11th edition of #VWSCC on a high note after a fantastic workshop led by @grimmelab.bsky.social. The recordings of the talks will be available on our website soon. ;-)
See you all next year!
#CompChem
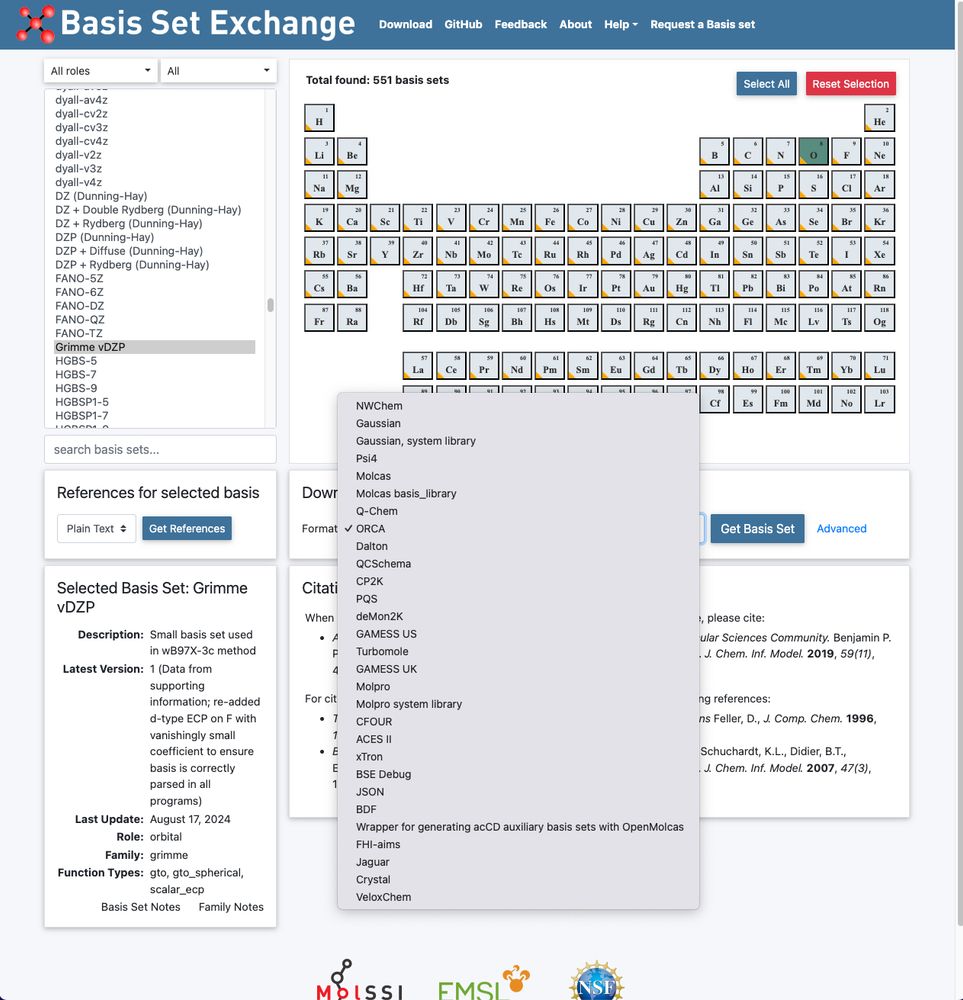
Our vDZP basis set utilized in the ⍵B97X-3c composite DFT method is now also available via www.basissetexchange.org (API-based: github.com/MolSSI-BSE/b...). 🎉
Many thanks to @Susi Lehtola & coworkers for jointly providing it there!
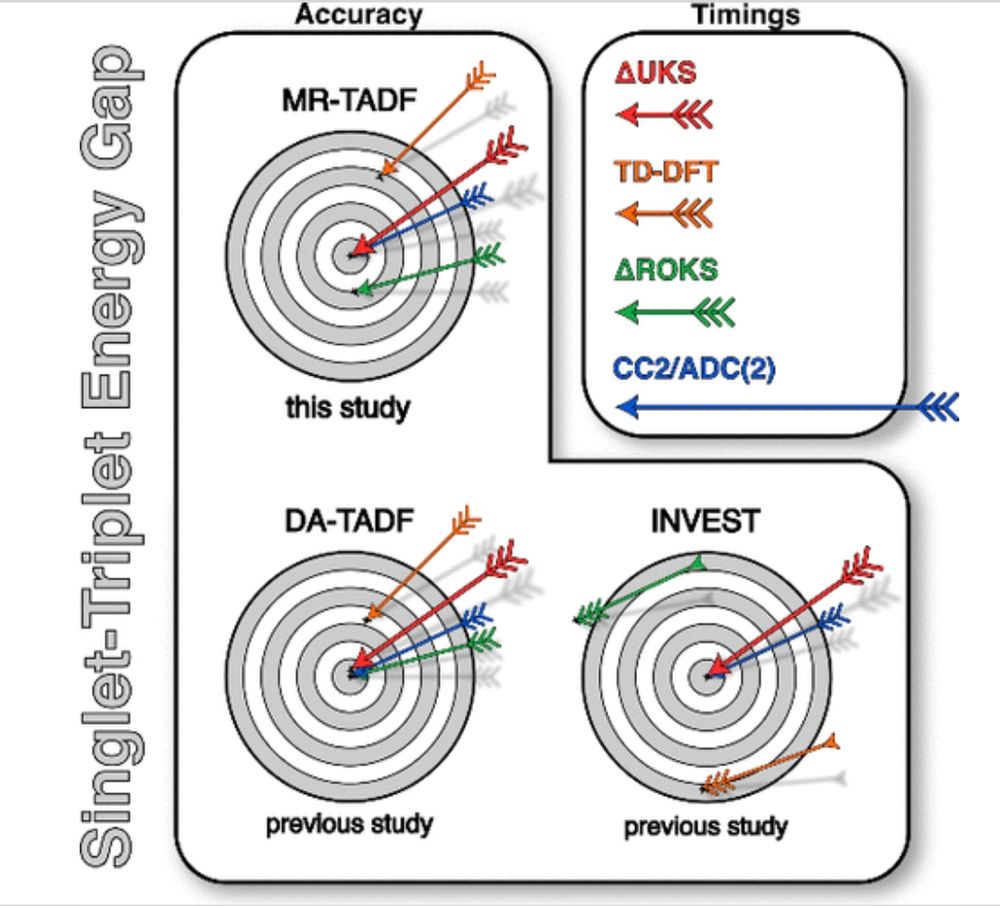
Wave function methods like CC2/ADC(2) are accurate (also some DHDFs), but too slow to cover large search spaces in high throughout screenings.
This is where the state-specific ΔDFT/UKS developed in cooperation with @grimmelab.bsky.social really shines
(same for classic DA-TADF and INVEST emitters):

I have created a timeline of semiempirical methods in quantum chemistry. Any thoughts, suggestions, or remarks on it? 💡 Have we missed anything?
22.01.2025 12:22 — 👍 19 🔁 2 💬 4 📌 1
Julia and her PI Stefan Grimme stand on either side of a statue of August Kekulé. Julia wears a traditional handcrafted PhD hat.
Julia from @grimmelab.bsky.social successfully defended her #PhDthesis this week. Congratulations! Julia worked on the "Development of #QuantumChemistry based Workflows for the Theoretical Description of #OrganicElectronics".
#ProudPI #PhDone
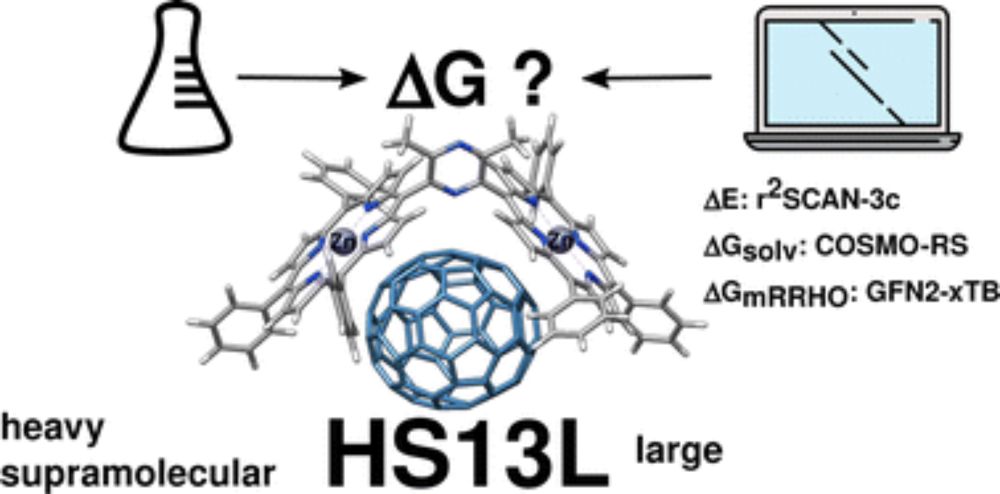
If you are interested in computing supramolecular complexes, take a look at this article in @PCCP
and see how well CREST and CENSO work for this challenging task: doi.org/10.1039/D2CP... @grimmelab.bsky.social
#compchem
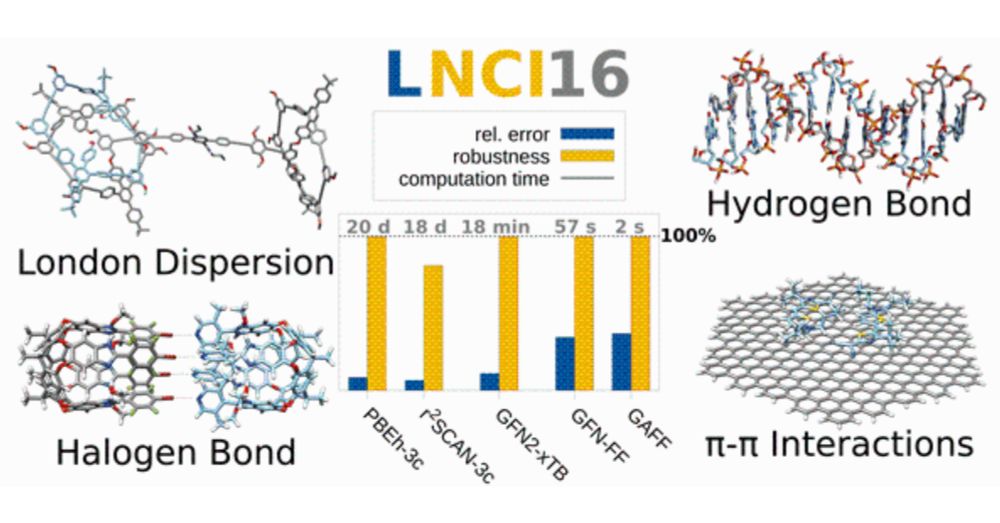
See how well low-cost methods describe non-covalent interactions in very large complexes (up to 2000 atoms!) and test your own methods on the LNCI16 benchmark set presented in our article @synlettjournal.bsky.social doi.org/10.1055/s-00... @grimmelab.bsky.social
17.01.2025 16:11 — 👍 13 🔁 4 💬 0 📌 0
Working with Stefan has always been an inspiring pleasure. It's particularly fascinating how naturally he navigates the manifold physics that govern chemistry, always on the look for a more elegant and efficient approximation.
This award is well deserved! Congratulations @grimmelab.bsky.social!
We're delighted to announce Stefan Grimme @grimmelab.bsky.social as the recipient of the 2025 Chemistry Europe Award! He'll be recognized "for his groundbreaking contributions that have had a profound impact on the scientific community” at #IUPAC2025.
www.chemistryviews.org/stefan-grimm...
#Chemsky

Stefan Grimme receives the 2025 Chemistry Europe Award!
Learn more at https://buff.ly/3BXDVhO.
#ChemistryAward #ChemistryEurope
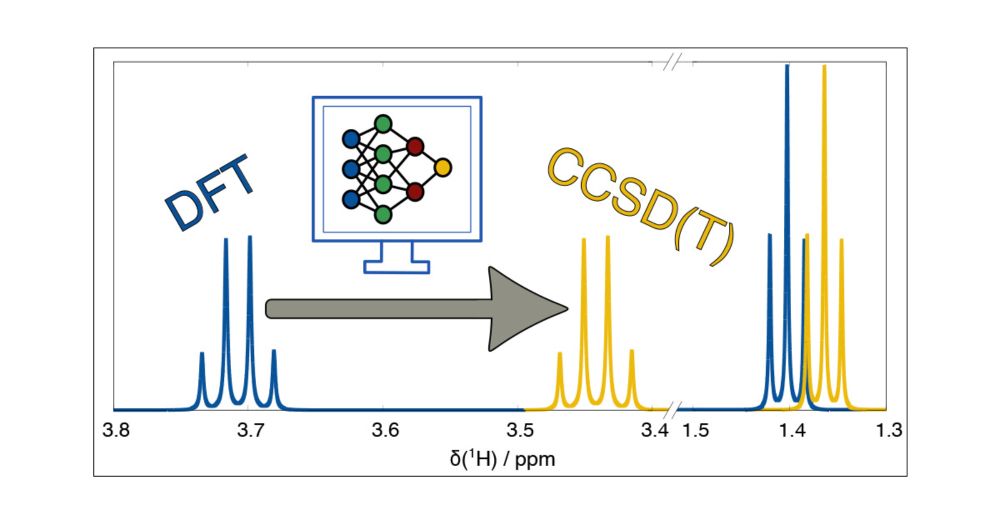
Take a look at Julius Kleine-Büning's work (from @grimmelab.bsky.social) work on ML-based corrections. IIRC, this massively improves the chemical shifts of 1H spectra. The software should be available online. pubs.acs.org/doi/abs/10.1...
08.01.2025 08:16 — 👍 14 🔁 5 💬 1 📌 0