Documentation of results rendered as of Tue Jan 6 19:25:41 2026
Data, code, and interactive visualizations for comparing amino-acid preferences across H3, H5, and H7 available at: jbloomlab.github.io/ha-preferenc...
Thanks to @jahn0.bsky.social for leading this with me, and also @bdadonaite.bsky.social, Caelan Radford, and @jbloomlab.bsky.social!
21.01.2026 19:22 — 👍 1 🔁 0 💬 0 📌 0
This also highlights limitation of using experimental measurements derived from a single genetic background for viral surveillance and vaccine immunogen design. Deep mutational scanning can be useful for predicting mutation effects in closely related variants, but less so across divergent homologs.
21.01.2026 19:22 — 👍 2 🔁 0 💬 1 📌 0
Overall, these results consistent with evolutionary contingency. Mutations can modify constraints at other sites, which snowballs over time.
21.01.2026 19:22 — 👍 2 🔁 0 💬 1 📌 0

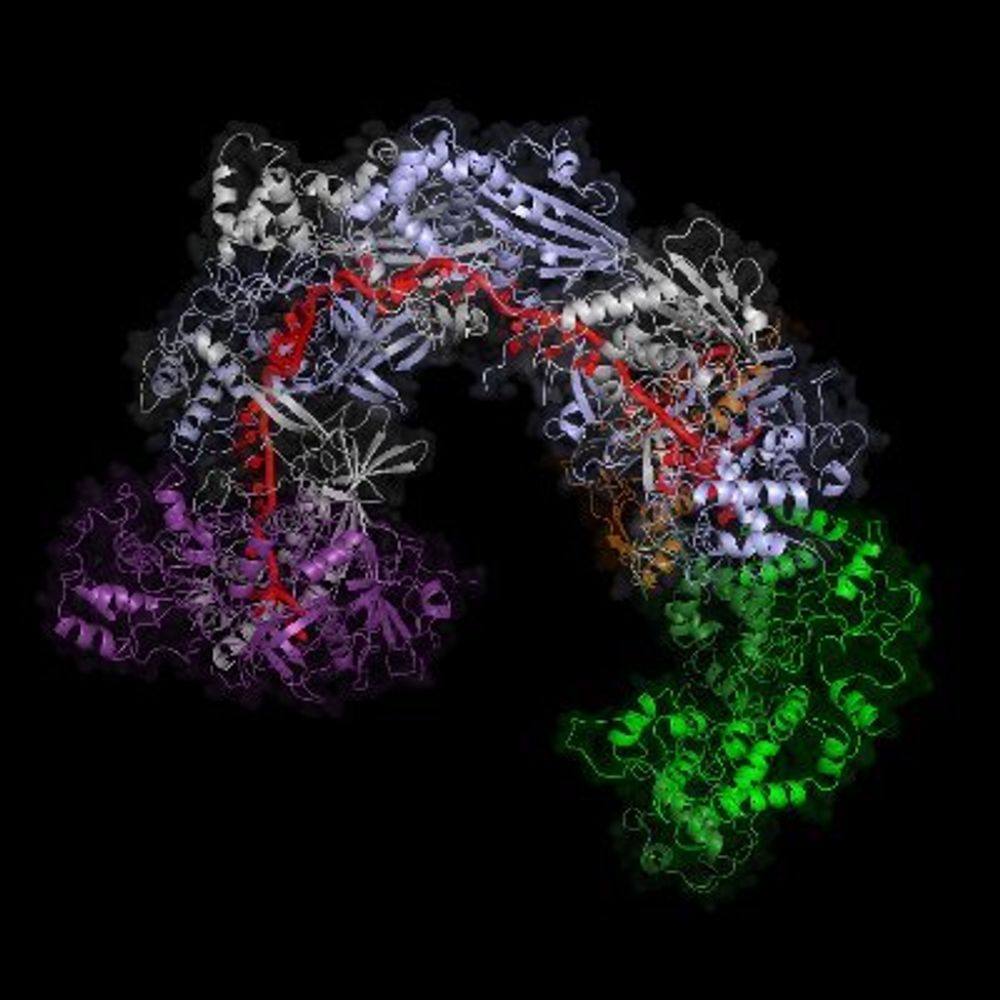
One example is site 176. H5/H7 tolerate similar amino acids, but both are sharply diverged from H3 which only tolerates positively charged K. Structure shows how contacting sites form constrained hydrogen bond network in H3, but same sites have been rewired into hydrophobic environment in H5/H7.
21.01.2026 19:22 — 👍 1 🔁 0 💬 1 📌 0
What explains sites with divergent amino-acid preferences? We find that they tend to be buried in the protein and have biochemically distinct wildtype amino acids in the subtypes.
21.01.2026 19:22 — 👍 2 🔁 0 💬 1 📌 0

~50% of sites display significant divergence in amino-acid preferences between HAs. HA2 domain of H3/H7 is noticeably less divergent, consistent with higher amino-acid conservation.
21.01.2026 19:22 — 👍 2 🔁 0 💬 1 📌 0

We then compared the H7 measurements to previously generated data for H5 (journals.plos.org/plosbiology/...) and H3 (www.nature.com/articles/s41...).
High divergence in amino-acid preferences = HA subtypes tolerate distinct amino acids (ex. site 86).
21.01.2026 19:22 — 👍 2 🔁 0 💬 1 📌 0
We first used pseudovirus deep mutational scanning to measure how all mutations to a recent H7 HA affect cell entry. This approach uses virions that can only undergo one round of cell entry and are therefore not capable of causing disease.
21.01.2026 19:22 — 👍 2 🔁 0 💬 1 📌 0

Epistatic drift causes gradual decay of predictability in protein evolution
The effect and fate of most mutations gradually become unpredictable as proteins evolve.
During protein evolution, mutation effects become less correlated as homologs diverge (www.science.org/doi/10.1126/...).
For HA, we wondered how sequence divergence on a nearly fixed structural backbone affects tolerance to further mutations.
21.01.2026 19:22 — 👍 3 🔁 0 💬 1 📌 0

As background, there are at least 19 influenza A virus HA subtypes. Many subtypes are highly diverged at the sequence level (~40% amino-acid identity), but protein structure and cell entry function are highly conserved.
21.01.2026 19:22 — 👍 2 🔁 0 💬 1 📌 0
Influenza hemagglutinin subtypes have different sequence constraints despite sharing extremely similar structures
Hemagglutinins (HA) from different influenza A virus subtypes share as little as ∼40% amino acid identity, yet their protein structure and cell entry function are highly conserved. Here we examine the extent that sequence constraints on HA differ across three subtypes. To do this, we first use pseudovirus deep mutational scanning to measure how all amino-acid mutations to an H7 HA affect its cell entry function. We then compare these new measurements to previously described measurements of how all mutations to H3 and H5 HAs affect cell entry function. We find that ∼50% of HA sites display substantially diverged preferences for different amino acids across the HA subtypes. The sites with the most divergent amino-acid preferences tend to be buried and have biochemically distinct wildtype amino acids in the different HA subtypes. We provide an example of how rewiring the interactions among contacting residues has dramatically shifted which amino acids are tolerated at specific sites. Overall, our results show how proteins with the same structure and function can become subject to very different site-specific evolutionary constraints as their sequences diverge. ### Competing Interest Statement JDB consults for Apriori Bio, Invivyd, Pfizer, GSK, and the Vaccine Company. JDB and BD are inventors on Fred Hutch licensed patents related to the deep mutational scanning of viral proteins. National Institute of Allergy and Infectious Diseases, R01AI165821, 75N93021C00015 U.S. National Science Foundation, DGE-2140004 Howard Hughes Medical Institute, https://ror.org/006w34k90
In new work by @jahn0.bsky.social and I in @jbloomlab.bsky.social, we investigate how sequence constraints differ across influenza HA subtypes.
We find ~50% of sites in HA display substantially different amino-acid preferences across H3, H5, and H7.
doi.org/10.64898/202...
21.01.2026 19:22 — 👍 23 🔁 10 💬 1 📌 0
Documentation of results rendered as of Tue Jan 6 19:25:41 2026
Data, code, and interactive visualizations for comparing amino-acid preferences across H3, H5, and H7 available at: jbloomlab.github.io/ha-preferenc...
Thanks to @jahn0.bsky.social for leading this with me, and also @bdadonaite.bsky.social, Caelan Radford, and @jbloomlab.bsky.social!
21.01.2026 18:58 — 👍 0 🔁 0 💬 0 📌 0
This also highlights limitation of using experimental measurements derived from a single genetic background for viral surveillance and vaccine immunogen design. Deep mutational scanning can be useful for predicting mutation effects in closely related variants, but less so across divergent homologs.
21.01.2026 18:58 — 👍 0 🔁 0 💬 1 📌 0
Overall, these results consistent with evolutionary contingency. Mutations can modify constraints at other sites, which snowballs over time.
21.01.2026 18:58 — 👍 0 🔁 0 💬 1 📌 0
One example is site 176. H5/H7 tolerate similar amino acids, but both are sharply diverged from H3 which only tolerates positively charged K. Structure shows how contacting sites form constrained hydrogen bond network in H3, but same sites have been rewired into hydrophobic environment in H5/H7.
21.01.2026 18:58 — 👍 0 🔁 0 💬 1 📌 0
What explains sites with divergent amino-acid preferences? We find that they tend to be buried in the protein and have biochemically distinct wildtype amino acids in the subtypes.
21.01.2026 18:58 — 👍 0 🔁 0 💬 1 📌 0
~50% of sites display significant divergence in amino-acid preferences between HAs. HA2 domain of H3/H7 is noticeably less divergent, consistent with higher amino-acid conservation.
21.01.2026 18:58 — 👍 0 🔁 0 💬 1 📌 0
We then compared the H7 measurements to previously generated data for H5 (journals.plos.org/plosbiology/...) and H3 (www.nature.com/articles/s41...).
High divergence in amino-acid preferences = HA subtypes tolerate distinct amino acids (ex. site 86).
21.01.2026 18:58 — 👍 0 🔁 0 💬 1 📌 0
We first used pseudovirus deep mutational scanning to measure how all mutations to a recent H7 HA affect cell entry. This approach uses virions that can only undergo one round of cell entry and are therefore not capable of causing disease.
21.01.2026 18:58 — 👍 0 🔁 0 💬 1 📌 0
Epistatic drift causes gradual decay of predictability in protein evolution
The effect and fate of most mutations gradually become unpredictable as proteins evolve.
During protein evolution, mutation effects become less correlated as homologs diverge (www.science.org/doi/10.1126/...).
For HA, we wondered how sequence divergence on a nearly fixed structural backbone affects tolerance to further mutations.
21.01.2026 18:58 — 👍 0 🔁 0 💬 1 📌 0
As background, there are at least 19 influenza A virus HA subtypes. Many subtypes are highly diverged at the sequence level (~40% amino-acid identity), but protein structure and cell entry function are highly conserved.
21.01.2026 18:58 — 👍 0 🔁 0 💬 1 📌 0
Microbes & mucus 🤩 | Gut Microbial Ecology | Wageningen University, NL
HFSP postdoctoral fellow @TypasLab @EMBLHeidelberg | Alumnus of @SorekLab @WeizmannScience. Interested in microbial interactions and warfare, microbial genomics & phages 🦠🧬👩🏽🔬
Research fellow at the Cancer Science Institute of Singapore | Cancer immunology focusing on antibodies and B cells in cancer | PhD @ UIUC | https://timothyjtan.github.io
Scientist + Humanist + Pugilist.
"Tip your hat; pop the chain; short Joe Louis; then wipe his nose with the hook. It's that simple." (c) Brother Naazim Richardson
https://linktr.ee/chike98
Associate Professor, MIT
Still thinking about the 10^9 mutations generated in your microbiome today.
Website: http://lieberman.science
News from the Sternberg lab at Columbia University, Howard Hughes Medical Institute.
Posts are from lab members and not Samuel Sternberg unless signed SHS. Posts represent personal views only.
Visit us at www.sternberglab.org
PhD candidate at UW Seattle Molecular and Cellular Biology program 🧬 Studying retinal development and disease 👁️
Cofounder/CEO Octant
BoD Ginkgo Bioworks
Defense Science Board for Emerging Biotech
Fmr: Associate Professor, UCLA
Evolutionary Biologist at Stanford. Rapid Evolution, Adaptation, and Genomics. Open Science advocate.
Discover the Languages of Biology
Build computational models to (help) solve biology? Join us! https://www.deboramarkslab.com
DM or mail me!
Evolving better E. coli for 75,000 generations. Prof at MSU, but opinions my own. (Ok, I also speak for billions -- er, TRILLIONS -- of E. coli.)
Website for LTEE: the-ltee.org
Banner pic from NYC, shared by Darwin. (The microbiologist, not the other one.)
Professor at Michigan State University. Trying to understand how the universe works, including people and animals. And plants and microbes. So, pretty much everything. (he/him)
We invest in scientists at all career stages who make discoveries that advance human health for decades to come.
Asst. Prof. of Medicine in GI at UC San Diego. Man of cultures (bacterial). Spatial Metagenomics & Multiplexed Assays. #Microbiome #SyntheticBiology
www.urtecholab.org
Assistant Prof. @uoknightcampus.bsky.social. Cofounder @synplexity.bsky.social. Gene synthesis, synbio, protein engineering, nanopores, multiplex assays. Opinions my own. www.plesalab.org
Evolutionary biochemistry at UChicago. Reconstructing ancient proteins.
Assistant Professor, University of Washington, Genome Sciences.
Previous: JSMF Fellow, Berkeley EECS
♡: Computational biology, evolutionary dynamics, quantitative immunology
https://dewitt-lab.github.io/
[disclaimer: opinions mine] 🇺🇸🚫👑
HHMI Freeman Hrabowski Scholar and Assistant Professor in the Department of Microbiology at the University of Washington studying host-pathogen interactions in beautiful Seattle.
(he/him). Opinions are my own and do not reflect those of my employer.
Theoretical biophysics. ENS-PSL / CNRS