If you ever need to fuzzy search some DNA, sassy is your tool.
Please spread the word; I think many people just outside my own circle could benefit from this :)
cc @rickbitloo.bsky.social
github.com/RagnarGrootK...
10.12.2025 15:50 — 👍 40 🔁 24 💬 4 📌 0

Output of `sassy grep -p ACTGGCATGAGAACTGAG -k1 human-genome.fa`. Should fuzzy matches with up to one error, grouped by file, then by record. The matching part is highlighted with colours (green=match, orange=mismatch, red=delete, blue=insert), and the strandedness of each match, the location, and the cost are shown.
Following ish's `filter` and bqtools' `grep`, Sassy now also has initial support for grep and filter!
Grep mode shows all matches, grouped per record, and is meant for human consumption.
Filter mode prints full matching (or non-matching) records to stdout or output files.
30.10.2025 23:46 — 👍 15 🔁 5 💬 1 📌 2
Was just checking if we could add the adapter search directly, then "slice" out all sub-reads so splitting does still look at the barcodes. And thanks for the discussions, nice to get ideas from what else to improve :)
25.10.2025 14:14 — 👍 2 🔁 0 💬 0 📌 0

Indeed does happen, but more often they are duplex reads that are not detected as such by Dorado. Then the sequences on either side of the mid strand adapter are reverse complement (example pic)
25.10.2025 14:12 — 👍 0 🔁 2 💬 1 📌 0
With Flye? Aah in the preprint we actually used Flye with Dorado trimmed reads 😅. You can see a list of "contaminated" assemblies here: zenodo.org/records/1739..., still quite a few, and this is very strict filtering. In reality there will be more
25.10.2025 09:03 — 👍 0 🔁 0 💬 0 📌 0
Yeah Porechop is probably "necessary" when using Dorado, though it shouldn't be. "Appreciable" heavily depends on the application. For assembly a few reads wrong is not always an issue, but for diagnostics a few reads wrong can be a big issue. Of course one should also check scores then, etc.
24.10.2025 14:27 — 👍 4 🔁 0 💬 1 📌 0

Hey Misha, on a 10K read sample around 60% is the expected dual-end (top), but often a single-end is enough to assign already. That puts it at around 90% (top 3 in image). Double barcode ligations do happen (bottom) but not sure about the exact stats of bleeding because of that.
24.10.2025 14:01 — 👍 2 🔁 0 💬 0 📌 0
To add, you can then add the desired pattern to barbell filter where you put “cut” marks (<<, >>) such that only the long reads is kept and the short contam is cut off
24.10.2025 12:33 — 👍 1 🔁 0 💬 0 📌 0
Agree, Barbell will just give a "complex" pattern for those reads which are not added to the output by default. Though, if the reads are very long, and contain little contamination (i.e. long read + short concat read at the end) it might still be worth it including it for assembly
24.10.2025 12:16 — 👍 2 🔁 0 💬 1 📌 0
Yeah most of NCBI is down, or outdated at the moment. We will upload them to Zenodo
24.10.2025 12:12 — 👍 3 🔁 1 💬 0 📌 0
Hey Kevin, that comes down to the difference between demultiplexing correct and removing contamination. If you had a read with NB01-NB02--read--. Dorado will demux to NB01, and you can remove NB02 contamination using Porechop, but that does mean your read was still demultiplexed incorrect initially
24.10.2025 10:04 — 👍 3 🔁 0 💬 1 📌 0
Really exciting that the preprint on Barbell, a new demultiplexer, is finally out!
It's the first tool that builds on Sassy, the approximate-DNA-searching tool that @rickbitloo.bsky.social and myself developed earlier this year, specifically with this application in mind.
23.10.2025 21:28 — 👍 20 🔁 15 💬 2 📌 0
See the thread for a quick summary! bsky.app/profile/rick...
23.10.2025 20:17 — 👍 0 🔁 0 💬 0 📌 0
Barbell Resolves Demultiplexing and Trimming Issues in Nanopore Data https://www.biorxiv.org/content/10.1101/2025.10.22.683865v1
23.10.2025 19:47 — 👍 6 🔁 3 💬 1 📌 0
In the pre-print we also discuss a specific tagmentation pattern causing partial loss of the second barcode, a custom barcode scoring scheme, and more: www.biorxiv.org/content/10.1...
23.10.2025 20:16 — 👍 3 🔁 0 💬 0 📌 0

In Barbell we solve this by first annotating all the reads, and then detecting all patterns, which looks like this:
23.10.2025 20:16 — 👍 3 🔁 0 💬 1 📌 0

Why is this a problem? Remaining adapters/barcodes not only contaminated assemblies but also created "artificial" links in taxonomic annotation, to Enterobacteriaceae, and to contaminated assemblies in NCBI.
23.10.2025 20:16 — 👍 5 🔁 0 💬 1 📌 1

Many tools, including the widely used Dorado, almost always only detected the *first* occurrence, leaving the rest untrimmed. In the figure black lines indicate matches to adapters + barcodes in trimmed reads.
23.10.2025 20:16 — 👍 2 🔁 0 💬 1 📌 0

For rapid barcoding only ~83% of the reads contain the expected single barcode on the left. The rest? Barcodes on both sides (6.1%), two barcodes on the left side (3.5%), and so on.
23.10.2025 20:16 — 👍 2 🔁 0 💬 1 📌 0
Around 10% of your Nanopore reads (SQK-RBK114) are incorrectly trimmed. Here is why, and how our new tool Barbell solves it:
www.biorxiv.org/content/10.1...
Want to get started? github.com/rickbeeloo/b...
23.10.2025 20:16 — 👍 51 🔁 31 💬 3 📌 4
Now also on biorxiv :)
26.07.2025 19:15 — 👍 5 🔁 2 💬 1 📌 0
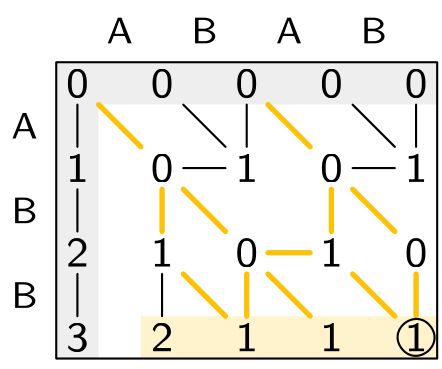
Sassy solves approximate string matching: finding all matches of a pattern in a text.
Sassy is out now!
Ever need to search for approximate matches of short DNA strings?
Sassy is the tool to use!
Available now wherever you get your code
With @rickbitloo.bsky.social
curiouscoding.nl/papers/sassy...
github.com/ragnarGrootK...
18.07.2025 20:20 — 👍 39 🔁 22 💬 2 📌 2
😂
10.07.2025 14:45 — 👍 0 🔁 0 💬 0 📌 0
Meta/viromics.
RNA virology, Archaeal viruses, Linux and FOSS.
Currently a postdoc at the Joint Genome Institute (formerly at TAU)
I love to code.
https://burntsushi.net
Scientist… pathogen genomics & antimicrobial resistance, computational biology & infectious disease epidemiology.
Co-Director LSHTM AMR Centre @lshtmamrcentre.bsky.social
holtlab.net | klebnet.org | typhoidgenomics.org | amr.lshtm.ac.uk
Research scientist interested in mosquitoes, infectious disease, nanopore seq and metagenomics. Views are my own.
Neophile. #ADHD. #rustlang at @datadoghq.com. #boardgames. Mercator Projection Apologist.
PhD candidate in Bioinformatics
(Environmental) Metagenomics, Phages
Inspired by Microbial Ecology and Evolution - Microbial Bioinformatics. Postdoc at Theoretical Biology and Bioinformatics (UU) & Bioinformatics (WUR)
postdoc at Viral Ecology and Omics group, @microverse.bsky.social @uni-jena.de. Phages, metagenomics, evolutionary modeling, microbial communities, kendo.
Bioinformatics Scientist at the Arc Institute.
Working at the intersection of functional genomics, systems biology, and machine learning. I also build rusty bioinformatics tools
https://github.com/noamteyssier
EUPHEM fellow/bioinformatician at RIVM, NL
Do my science @ace_uq studying coral reef microbiomes. Data wrangler, meta-omics and long-read wonk, clean energy enthusiast, Saganist zealot, collector of weird zoology facts, other nonsense.
Professor for Personalised Molecular Microbiology. University Medical Center Groningen, Isala Hospital Zwolle, and University of Utah. PhD in coronavirus cell interactions (1996). #metagenomics #metatransriptomics #AMR #artificialintelligence
Bioinformatics geek 🤓 crafting Rust-y tools 🦀 for microbial genomes 🦠 🧬.
Trying to master Dad mode 👨🍼
See what I'm up to here: https://github.com/mbhall88
Medical Molecular Microbiologist at the Amsterdam-UMC. Interested in bacterial genomics, antimicrobial resistance and bioinformatics.
Lecturer at UNSW Sydney; Visiting Scientist at Garvan Institute of Medical Research -
Designing embedded systems for bioinformatics applications.
Professor of Computer Science @ JHU. https://www.langmead-lab.org/ https://www.youtube.com/BenLangmead
Microbial ecologist, postdoc at FSU
Postdoc @ DOE JGI Berkeley Lab 🇺🇸 | Former PhD Student @ Flinders University 🇦🇺 | #bioinformatics #phage #microbiome 🦠💻🧬 | she/her
https://github.com/susiegriggo