In Edinburgh for the RSC/SCI Symposium on Kinase Inhibitor Design—excited to see @jessbwhite.bsky.social share her work on the missense kinase toolkit! #Kinase2025
13.05.2025 10:51 — 👍 4 🔁 0 💬 0 📌 1
@markpolk.io.bsky.social
I use robots and biophysics to study cancer therapies in the Chodera Lab at Memorial Sloan Kettering Cancer Center. Views are my own.
In Edinburgh for the RSC/SCI Symposium on Kinase Inhibitor Design—excited to see @jessbwhite.bsky.social share her work on the missense kinase toolkit! #Kinase2025
13.05.2025 10:51 — 👍 4 🔁 0 💬 0 📌 1
the longest journey begins with a single step. luckily for us, @arianaclerkin.bsky.social is writing hers down!
read the first PLUMB blab of many below, and follow along as Ariana builds an open comp.chem project from the ground up.
Check out our recent preprint 👇on counting particles to estimate populations in #cryoem: noise can bias the estimates; with Luke Evans & a great team.
30.03.2025 13:29 — 👍 29 🔁 13 💬 0 📌 1
Five people, covered in bright Holi powder, stand arm-in-arm in front of a waterfront railing with a cityscape in the background. They’re all smiling and wearing colorful, powder-stained clothes.
Happy Holi, from the @jchodera.bsky.social lab!!
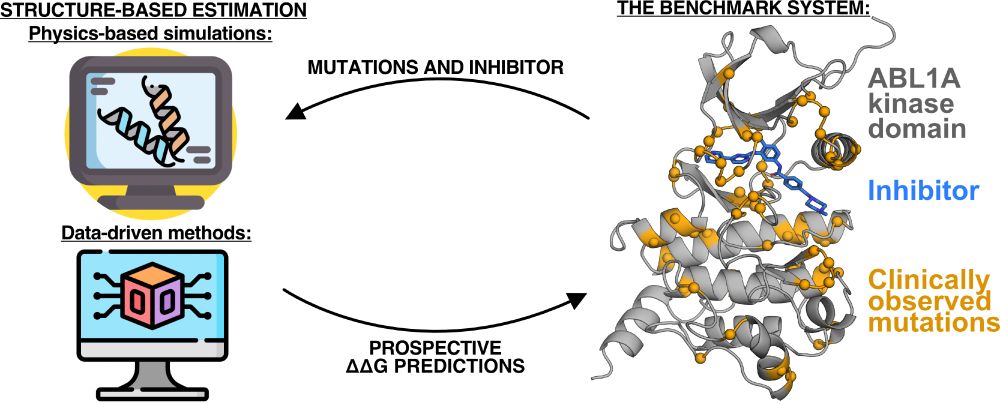
14.03.2025 21:51 — 👍 27 🔁 2 💬 0 📌 0This work is now published in J Phys Chem B! Check out our work showing that simulations predict the impact of distal mutations on kinase-inhibitor binding, and our experimental NanoBRET dataset of 94 kinase mutations that provide a benchmark for future methods. Link:
pubs.acs.org/doi/full/10....
"The #undergraduates usually bring samples from the projects they are working on. In this case, the data they obtain from this trip may become the last piece of data they need before submitting their #research for #publication"🥳🥳 @actacrystc.iucr.org @actacryste.iucr.org #crystallography #education
19.02.2025 22:40 — 👍 4 🔁 2 💬 0 📌 0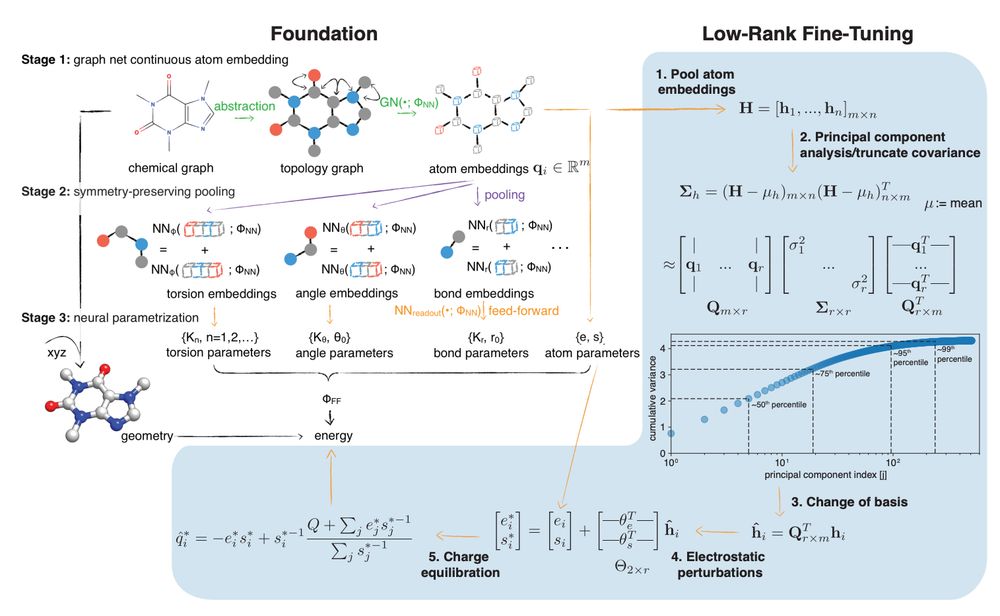
Figure 1 from arXiv preprint https://doi.org/10.1101/2025.01.06.631610 Fig. 1 Espaloma is an end-to-end differentiable molecular mechanics parameter assignment scheme for arbitrary organic molecules. Espaloma (extensible surrogate potential optimized by message-passing) is a modular approach for directly computing molecular mechanics force field parameters FFF from a chemical graph G such as a small molecule or biopolymer via a process that is fully differentiable in the model parameters FNN. In Stage 1, a graph neural network is used to generate continuous latent atom embeddings describing local chemical environments from the chemical graph. In Stage 2, these atom embeddings are transformed into feature vectors that preserve appropriate symmetries for atom, bond, angle, and proper/improper torsion inference via Janossy pooling.54 In Stage 3, molecular mechanics parameters are directly predicted from these feature vectors using feed-forward neural networks. This parameter assignment process is performed once per molecular species, allowing the potential energy to be rapidly computed using standard molecular mechanics or molecular dynamics frameworks thereafter. The collection of parameters FNN describing the espaloma model can be considered as the equivalent complete specification of a traditional molecular mechanics force field such as GAFF38,39/AM1-BCC55,56 in that it encodes the equivalent of traditional typing rules, parameter assignment tables, and even partial charge models. Reproduced from ref. 49 with permission from the Royal Society of Chemistry.
Everything is chaos, but I wanted to share some awesome recent science from the lab that hints at where the future of biomolecular simulation is headed:
Foundation simulation models that can be fine-tuned to experimental free energy data to produce systematically more accurate predictions.
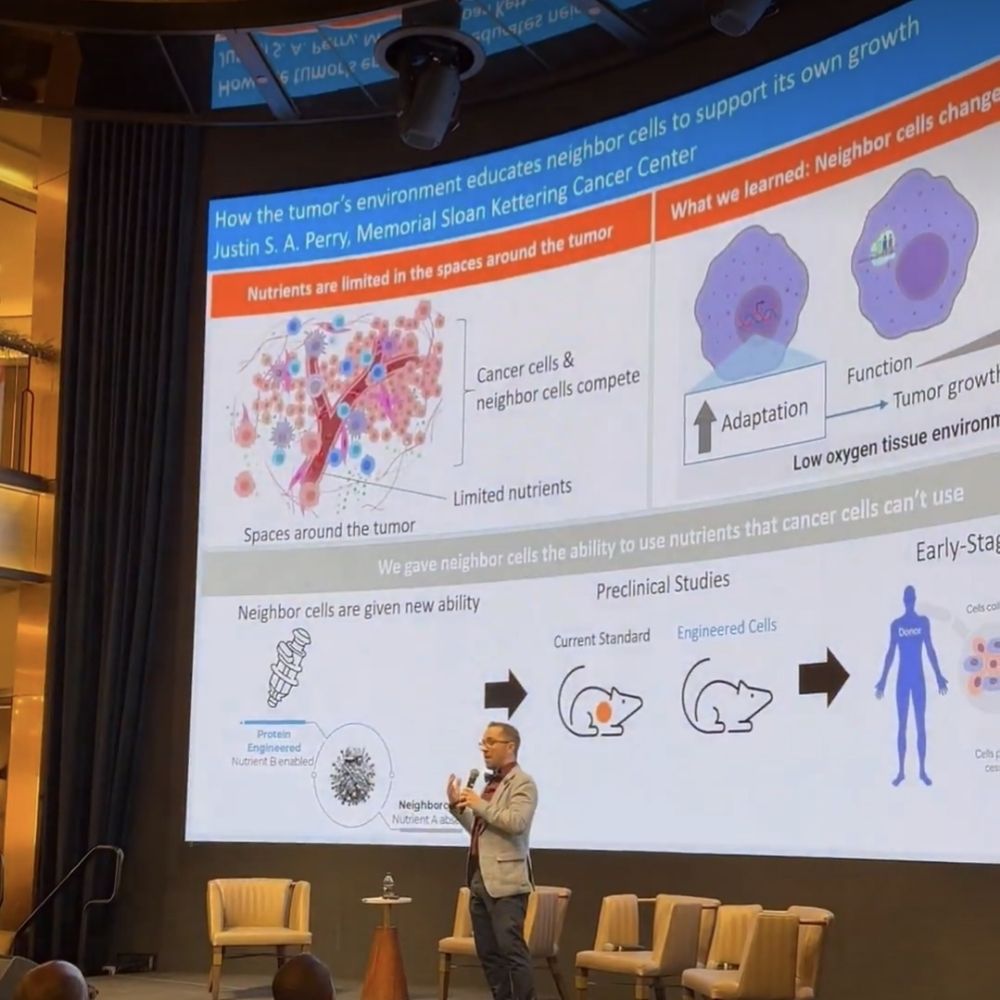
I'll be presenting my work prospectively studying clinical mutations and their impact on kinase inhibitor binding at #bps2025! Come see my talk on Wednesday morning @ 8:45am.
16.02.2025 18:53 — 👍 35 🔁 9 💬 1 📌 0Also from the @jchodera.bsky.social lab: my phenomenal mentor @sukritsingh92.bsky.social will give a talk on mechanism-based modeling of drug resistant mutations in cancer in room 515A on Wednesday at 8:45 AM.
16.02.2025 18:47 — 👍 4 🔁 0 💬 0 📌 0Excited to be in LA for #BPS2025 @biophysicalsoc.bsky.social! I’ll be presenting a poster on our high-throughput fluorescence-based assay for kinase inhibitor binding in the West Exhibit Hall on Monday at 2:45 PM (board B60).
16.02.2025 18:47 — 👍 8 🔁 4 💬 1 📌 1Also from the @jchodera.bsky.social lab: my phenomenal mentor @sukritsingh92.bsky.social will give a talk on mechanism-based modeling of drug resistant mutations in cancer on Wednesday at 8:45 AM in room 515A.
16.02.2025 18:44 — 👍 0 🔁 0 💬 0 📌 0
I'm at @biophysicalsoc.bsky.social #BPS2025 with a bunch of folks from the Structural and Molecular Biophysics @flatironinstitute.org group-- come say hi and check out our posters/talks! @sonyahanson.bsky.social @pilarcossio.bsky.social @miroastore.bsky.social
15.02.2025 21:54 — 👍 9 🔁 5 💬 0 📌 0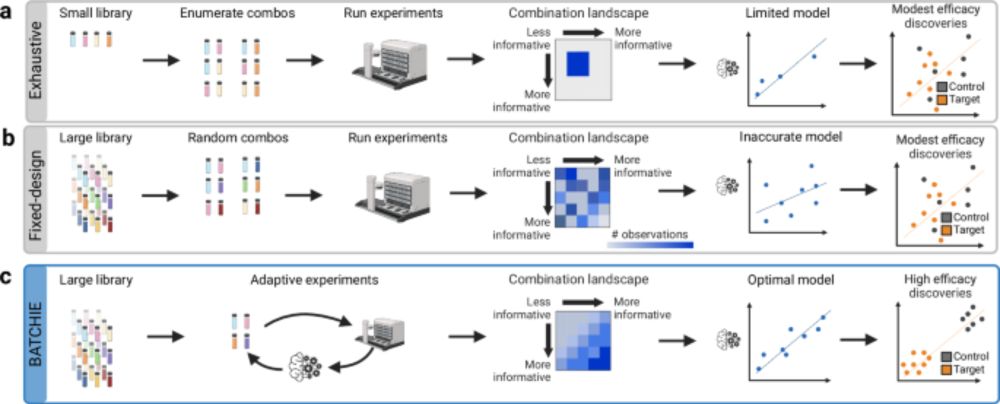
Happy to have been a member of the BATCHIE team who worked on this recent publication featured by @MSKLibrary: A Bayesian active learning platform for scalable combination drug screens dx.doi.org/10.1038/s414...
14.01.2025 15:04 — 👍 11 🔁 1 💬 0 📌 0Happy new year!
2024 was a productive year, and 2025 stands to be even more exciting!
Our thanks to everyone who helped, and some reflections: foldingathome.org/2025/01/09/h...

I’m standing on a walkway in the Diamond Light Source facility. The hall I am standing in is in the shape of a ring, and a portion of it — with exposed ducts and pipes — is visible behind me. A yellow beamline station can be seen behind me, on a level below the one I am on.
So grateful to have had the opportunity to chat science in Berlin with our collaborators in the Volkamer Lab and in the UK at the Oxford Centre for Medicines Discovery and the Diamond Light Source. Many thanks to DLS for the tour!
10.12.2024 17:12 — 👍 5 🔁 1 💬 0 📌 0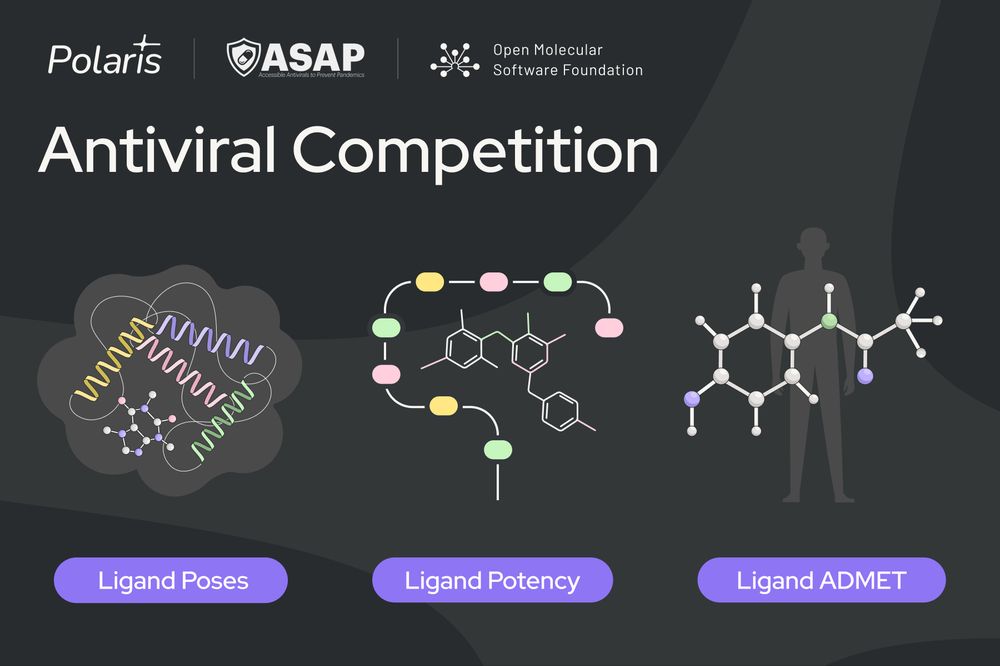
🦠 We’re excited to announce our first competition in partnership with @asapdiscovery.bsky.social and @omsf.io!
Test your skills across three sub-challenges revolving around SARS-CoV-2 and MERS-CoV Mpro🧵
Full details: polarishub.io/competitions
Blog: polarishub.io/blog/antivir...
Automated plate handling in the Chodera Lab, part of our effort to study kinase inhibitors in high throughput.
27.11.2024 16:44 — 👍 19 🔁 4 💬 0 📌 2