Phold's manuscript is now available @narjournal.bsky.social thanks to @susiegriggo.bsky.social @npbhavya.bsky.social @vijinim.bsky.social @linsalrob.bsky.social @martinsteinegger.bsky.social @milot.bsky.social @eunbelivable.bsky.social & others not on bsky #phagesky academic.oup.com/nar/article/...
14.01.2026 05:10 — 👍 82 🔁 44 💬 1 📌 1
P2 Solo announcement and the trade-offs of a more stable ONT
a blog for miscellaneous bioinformatics stuff
New blog post with some thoughts on @nanoporetech.com and their recent announcement that the P2 Solo will be discontinued:
rrwick.github.io/2026/01/21/p...
21.01.2026 03:38 — 👍 21 🔁 14 💬 0 📌 0
Viewing tables on the command line
a blog for miscellaneous bioinformatics stuff
Just updated this old blog post:
rrwick.github.io/2023/04/19/v...
Now uses qsv instead of xsv, plus some other miscellaneous tweaks/fixes.
In the 2+ years since I wrote this post, I use my tv function a LOT.
13.01.2026 01:09 — 👍 5 🔁 0 💬 0 📌 0
And since both metaMDBG and Myloasm had new versions after the paper was accepted, here's a blog post with an updated benchmark:
rrwick.github.io/2025/09/23/a...
(3/3)
29.09.2025 04:11 — 👍 2 🔁 0 💬 0 📌 0
Here's the ultra-short version:
If you want the best possible long-read bacterial genome assemblies, Autocycler is the tool for you! It is computationally intensive (due to the need to generate many alternative input assemblies) but consistently more accurate than other methods.
(2/3)
29.09.2025 04:11 — 👍 2 🔁 0 💬 1 📌 0
One caveat though: my experience is with isolates that (usually) have a single 'true' sequence to aim for. In a metagenome with natural variation (i.e. a mixture of multiple 'true' sequences), I'm not sure how Dorado/Medaka would behave...
24.09.2025 00:59 — 👍 1 🔁 0 💬 0 📌 0
I have limited experience with long-read metagenome assembly, so I don't have any special insight here. But I like the examples shown @floriantrigodet.bsky.social's preprint - it shows how sometimes one strange read (e.g. a chimera) can throw off the assembly.
23.09.2025 21:37 — 👍 0 🔁 0 💬 1 📌 0
No, the assemblies are not Dorado/Medaka polished. I predict doing so would significantly reduce error rates, especially for the higher-error assemblies - Dorado/Medaka is quite good at fixing small-to-medium scale errors. But for lower-error assemblies (e.g. Autocycler), it may not change much.
23.09.2025 21:36 — 👍 1 🔁 0 💬 1 📌 0
I agree, most real-world MAGs probably have more errors than isolates, especially if low depth. Also, metagenomes may have within-species variation, and then the ideal MAG is (arguably) some sort of consensus. Especially if there is structural variation, this can be a BIG challenge for assemblers.
23.09.2025 06:09 — 👍 2 🔁 0 💬 1 📌 0

Boxplots showing the benchmarking results from my blog post: old-vs-new versions of metaMDBG and Myloasm
23.09.2025 01:53 — 👍 4 🔁 0 💬 0 📌 0
Benchmark update: metaMDBG and Myloasm
a blog for miscellaneous bioinformatics stuff
New blog post!
metaMDBG (@gaetanbenoit.bsky.social) and Myloasm (@jimshaw.bsky.social) have had recent releases, so I updated the benchmarks from the Autocycler paper:
rrwick.github.io/2025/09/23/a...
Both tools improved considerably! Time to update your conda environments 😄
23.09.2025 01:53 — 👍 35 🔁 26 💬 4 📌 0
Preprint out for myloasm, our new nanopore / HiFi metagenome assembler!
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
07.09.2025 23:34 — 👍 114 🔁 80 💬 5 📌 5
Amazing, thanks! Will keep an eye out for this preprint.
07.09.2025 21:03 — 👍 1 🔁 0 💬 0 📌 0
I haven't yet, but that is absolutely something I should try. I should also more generally educate myself on best practices in telomere assembly, e.g. with that paper Adam linked. This is new to me!
07.09.2025 21:02 — 👍 1 🔁 0 💬 0 📌 0
Thanks for this - looks interesting!
In the paper you said: "...Illumina sequencing can generate spurious indels within HTs, especially for HT lengths longer than 14 bp." Do you have a sense of how bad this gets for really long homopolymers, e.g. 20+ bp?
05.09.2025 01:22 — 👍 0 🔁 0 💬 1 📌 0
I'm sure there are more robust ways to go about telomere assembly - I'm not very experienced with T2T eukaryote genome assemblies 😬
05.09.2025 01:10 — 👍 1 🔁 0 💬 0 📌 0
And my manual telomere fixing was makeshift. I pieced together a few assemblies (mostly from Flye) that extended all the way to the telomeres. And then I manually repaired the telomeres to be exact 6-mer repeats - i.e. I assumed any deviation from the 6-mer was ONT error not real biology.
05.09.2025 01:10 — 👍 1 🔁 0 💬 3 📌 0
Since all Illumina sequencing involves some PCR (bridge amplification), I also wonder at what length Illumina reads start to fail with homopolymers. Can they reliably sequence 20-mers? 40-mers? 60-mers? It's a hard question to answer if every sequencing tech struggles with these...
05.09.2025 01:10 — 👍 2 🔁 0 💬 3 📌 0
I too am wary. I suppose the hope is that by limiting changes to long homopolymers, polishing will fix more errors than it introduces. I.e. I'm guessing that ONT's long-homopolymer error rate is greater than cross-sample homopolymer differences. But this is very much unproven!
05.09.2025 01:10 — 👍 2 🔁 0 💬 1 📌 0
Cross-sample homopolymer polishing with Pypolca
a blog for miscellaneous bioinformatics stuff
New blog post!
I added a new feature to @gbouras13.bsky.social's Pypolca: homopolymer-only polishing. Potentially useful for cross-sample polishing - early test on Cryptosporidium looks promising.
Check it out here:
rrwick.github.io/2025/09/04/h...
04.09.2025 06:37 — 👍 18 🔁 7 💬 2 📌 1
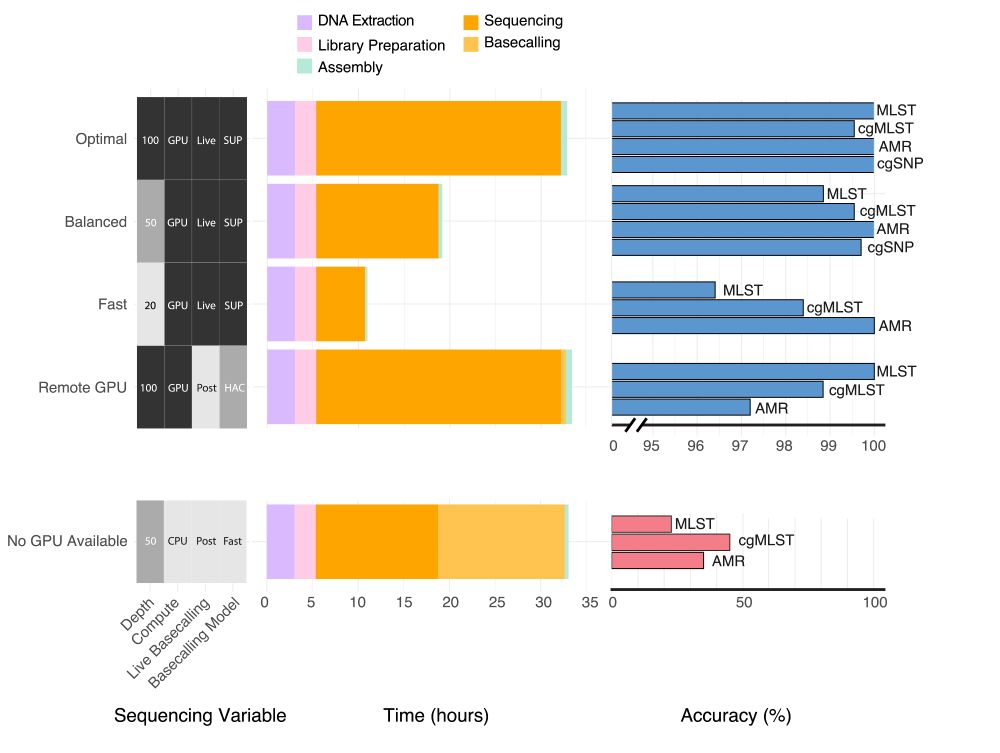
Pleased to say that our preprint benchmarking Nanopore data for MLST, cgMLST, cgSNP & AMR typing from bacterial isolates is out! TL;DR you can get almost perfect results from 50x depth using live SUP basecalling with a GPU in under 20 hours #microsky#IDsky 🦠🧬🖥️ /1
www.medrxiv.org/content/10.1...
30.07.2025 02:10 — 👍 46 🔁 29 💬 3 📌 2
However, I hear rumours that ONT might be working on a new move-table-aware bacterial polishing model. See my blog post from Feb for details: rrwick.github.io/2025/02/07/d.... If true, I'll be eager to test it out when released.
10.06.2025 02:37 — 👍 4 🔁 0 💬 0 📌 0
Release v1.0.1 · nanoporetech/dorado
[1.0.1] (4 June 2025)
This release introduces support in the --bacteria mode of Dorado polish for data basecalled with v5.2 models and improves the speed of 5mCG_5hmCG calling with v5.0 and v5.2 mo...
Minor new release of @nanoporetech.com's Dorado:
github.com/nanoporetech...
It supports using --bacteria when polishing an assembly with v5.2.0 data, which is nice! But if I understand correctly, it's the same bacterial polishing model from Sep 2024, not a new model.
10.06.2025 02:37 — 👍 8 🔁 1 💬 1 📌 0
Good to know - thanks for clarifying. Makes sense for a tool that's designed to work with big metagenomic datasets. I'm using it a bit out of its domain on a bacterial isolate.
30.05.2025 05:43 — 👍 0 🔁 0 💬 1 📌 0
The read set was only 240 MB (gzipped), so it is memory hungry. The myloasm docs do acknowledge that it uses more memory than other assemblers.
Also, I ran my tests on an ARM Mac, but the docs suggest that myloasm (specifically the polishing step) will be even faster on x86-64 CPUs with AVX2.
30.05.2025 02:43 — 👍 2 🔁 0 💬 1 📌 0
Just ran a few more tests through GNU time:
1 thread: 435 seconds, 10.1 GB RAM
2 threads: 238 seconds, 10.0 GB RAM
4 threads: 133 seconds, 10.1 GB RAM
8 threads: 73 seconds, 10.1 GB RAM
16 threads: 49 seconds, 13.3 GB RAM
30.05.2025 02:42 — 👍 2 🔁 0 💬 3 📌 0
I tested myloasm on a 50x Klebsiella isolate, and it was very fast - only took about 1 minute to complete (on my Macbook).
29.05.2025 05:01 — 👍 15 🔁 4 💬 2 📌 0
Professor at DTU NNF Center for Biosustainability with interest in bioactive compounds, comp. biol., WGS and much more; hobby photographer. Views are my own.
Fighting mental health stigma and toxic research culture. Proud to support https://twloha.com/.
And when life gives you lemons, make a G&T.
She/Her
Science and stories about our awesome planet!
Find us on Youtube: https://www.youtube.com/@MinuteEarth
Associate Professor
DFCI & HMS
Postdoc at Dana-Farber and Harvard Med with Heng Li (@lh3lh3.bsky.social). Prev: UBC / UofT.
I like thinking about biological sequence analysis and its applications to metagenomics / microbial genomics.
https://jim-shaw-bluenote.github.io
Research scientist interested in mosquitoes, infectious disease, nanopore seq and metagenomics. Views are my own.
Microbial genomics | Computational biology
Located at the Alfred Hospital/Monash Uni
Chief Data Scientist - Black Ochre Data Labs (blackochrelabs.au)
Past President - ABACBS (abacbs.org)
Adjunct A/Professor - Australian National University (anu.edu.au)
Interested in using multiomics to study complex diseases in Indigenous Australians
Bioinformatician @ MDU PHL, Melbourne
Enabling the Australian research community and industry access to nationally significant digital research infrastructure, platforms, skills and data collections. Enabled by #NCRIS. Visit https://ardc.edu.au/
Delivering world-class bioinformatics training in data-driven life sciences virtually and onsite at EMBL's European Bioinformatics Institute in Hinxton, UK.
www.ebi.ac.uk/training
EMBL's European Bioinformatics Institute (EMBL-EBI) provides open biological data resources and tools, and performs basic research in computational biology. https://www.ebi.ac.uk/
The Australian Bioinformatics and Computational Biology Society Inc. Promoting the science and profession of bioinformatics
https://www.abacbs.org
Bioinformatician, data scientist, software developer
Also @_lazappi_ and @lazappi@mastodon.au
Postdoc @ DOE JGI Berkeley Lab 🇺🇸 | Former PhD Student @ Flinders University 🇦🇺 | #bioinformatics #phage #microbiome 🦠💻🧬 | she/her
https://github.com/susiegriggo
Lead Bioinformatician in the Stroud Lab @UniMelb Bio21 Institute | Computational Mass Spec Multi-Omics | Chair of ABACBS Diversity, Inclusion and Accessibility Committee
Software, Hardware, Machine Learning Inference.
Postdoc researcher in bioinformatics at Pasteur institute. Scalable methods and software for metagenomics. https://github.com/GaetanBenoitDev