Back online. Not sure if it is a bug in my code or a hiccup at the hosting service.
01.10.2025 14:29 — 👍 1 🔁 0 💬 0 📌 0

Do you know ~60% of human SVs fall in ~1% of GRCh38? See our new preprint: arxiv.org/abs/2509.23057 and the companion blog post on how we started this project and longdust: lh3.github.io/2025/09/29/o.... Work with Alvin Qin
30.09.2025 02:19 — 👍 75 🔁 27 💬 0 📌 0
arXiv accepted our assembly review two years ago. That was written in MS Word, so PDF-only. Nonetheless, at that time they didn't require TeX source as I remember. Something might have been changed internally.
29.09.2025 01:26 — 👍 2 🔁 0 💬 0 📌 0
And learn what fully AI-generated websites look like. Avoid them, as they are more likely to be scam.
15.09.2025 12:18 — 👍 9 🔁 3 💬 1 📌 0
Heads up: ignore samtools dot org, similarly minimap2 dot com and likely others. It's owned by a known phishing site and while the binaries they offer look valid currently (but note they may be serving us different binaries to others), that could change.
Ie: it's not us (Samtools team)! Be warned
15.09.2025 08:40 — 👍 141 🔁 126 💬 2 📌 4

New blog post – A quick look at Roche's SBX
lh3.github.io/2025/09/11/a...
12.09.2025 03:26 — 👍 56 🔁 30 💬 2 📌 3
Now preprinted at arxiv.org/abs/2509.07357
10.09.2025 02:10 — 👍 21 🔁 7 💬 0 📌 0
Preprint out for myloasm, our new nanopore / HiFi metagenome assembler!
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
07.09.2025 23:34 — 👍 110 🔁 76 💬 5 📌 5
High-resolution metagenome assembly for modern long reads with myloasm https://www.biorxiv.org/content/10.1101/2025.09.05.674543v1
07.09.2025 04:47 — 👍 18 🔁 8 💬 0 📌 1
Of course, also thank Andrea Guarracino and Andrew Carroll for their quick and careful review!
04.09.2025 17:09 — 👍 3 🔁 0 💬 0 📌 0
"Received: July 4, 2025. Revised: August 7, 2025. Accepted: August 15, 2025" and published on September 4. This is a simple and straightforward paper, but the speedy editorial process is still impressive. It could have been even faster if I had responded the initial editorial request more timely.
04.09.2025 16:55 — 👍 10 🔁 1 💬 2 📌 1
Now published in GigaScience with minor improvements: academic.oup.com/gigascience/...
* Download: zenodo.org/records/1490...
* More info: github.com/lh3/panmask
04.09.2025 16:44 — 👍 30 🔁 10 💬 1 📌 1

(Harvard STAT115): Introduction to Bioinformatics and Computational Biology by Shirley Liu.
liulab-dfci.github.io/bioinfo-com...
27.08.2025 13:45 — 👍 14 🔁 3 💬 0 📌 0
Timing and practical needs are key factors. BCF also has a proper spec and an okay library and it is based on bgzf. The library is more complex to use because VCF is more complex.
07.08.2025 13:04 — 👍 2 🔁 0 💬 1 📌 0
CRAM also ticks some of the points, but the storage cost alone wins users over.
07.08.2025 13:01 — 👍 0 🔁 0 💬 0 📌 0
BAM is more of a literal dump. BCF is not used often because 1) VCF is a small fraction of SAM. Performance is not as critical. 2) Tabix is good enough. 3) Too complex to implement as VCF is not designed with binary in mind. 4) Too late. GATK started to support in 2019. 5) Binary version changes.
07.08.2025 12:49 — 👍 0 🔁 0 💬 3 📌 0
I think genomic file formats should be text-first and designed with binary representations in mind
06.08.2025 22:23 — 👍 4 🔁 0 💬 2 📌 0

Preprint on "Finding easy regions for short-read variant calling from pangenome data": arxiv.org/abs/2507.03718
08.07.2025 02:14 — 👍 31 🔁 13 💬 0 📌 1
Pangolin only supports SNPs and doesn't distinguish donor/acceptor but mutating cTaAt has little effect, too. I don't have alphaGenome numbers. Overall, this comes back to @ewanbirney.bsky.social's question: Is BP essential to splicing? Do these models really see BP?
29.06.2025 15:32 — 👍 0 🔁 0 💬 1 📌 0
chr3:143021319:cTaAt matches the yTnAy BP consensus at the 1-based coordinate. The Broad's SpliceAI server doesn't think chr3:143021320:TaA>CaG would lead to an acceptor loss – it's non-essential to splicing. SpliceAI thinks donor might be more affected.
29.06.2025 15:32 — 👍 0 🔁 0 💬 1 📌 0
Thanks for the explanation! This is a good example then. It will be interesting to see if pangolin or spliceAI can capture these with ISM. Another question is how often BPs are found across all acceptor sites.
26.06.2025 23:42 — 👍 0 🔁 0 💬 1 📌 0
IMHO, it is important to understand what a model learns; otherwise, the model might just captures nuances irrelevant to biology. 4/
26.06.2025 02:54 — 👍 4 🔁 0 💬 1 📌 0
Generally, my intuition is large DNNs draw power from compositional differences between introns and exons. For example, if we see a transition from 40% GC to 60% GC, a GT in the middle is more likely to be a real donor. I don't know how much signals DNNs learn just from splice motifs. 3/3
26.06.2025 02:51 — 👍 2 🔁 0 💬 1 📌 0
Fig 3b deletes 4bp right after a donor GT on the minus strand. This reduces minisplice log-odd from 9 to -1. It is also a trivial case. I am more interested in cases where a SNP >20-50bp away from splice site may affect splicing, with biological evidence. 2/3
26.06.2025 02:51 — 👍 0 🔁 0 💬 1 📌 0
They speculate it is a branch point at one particular acceptor in Fig 3d. I am not sure if that is the real one. Similarly, the exon motif seems a guess as well. Also, Fig 3c changes the G in GT. This of courses kills the splice site. 1/3
26.06.2025 02:51 — 👍 0 🔁 0 💬 2 📌 0
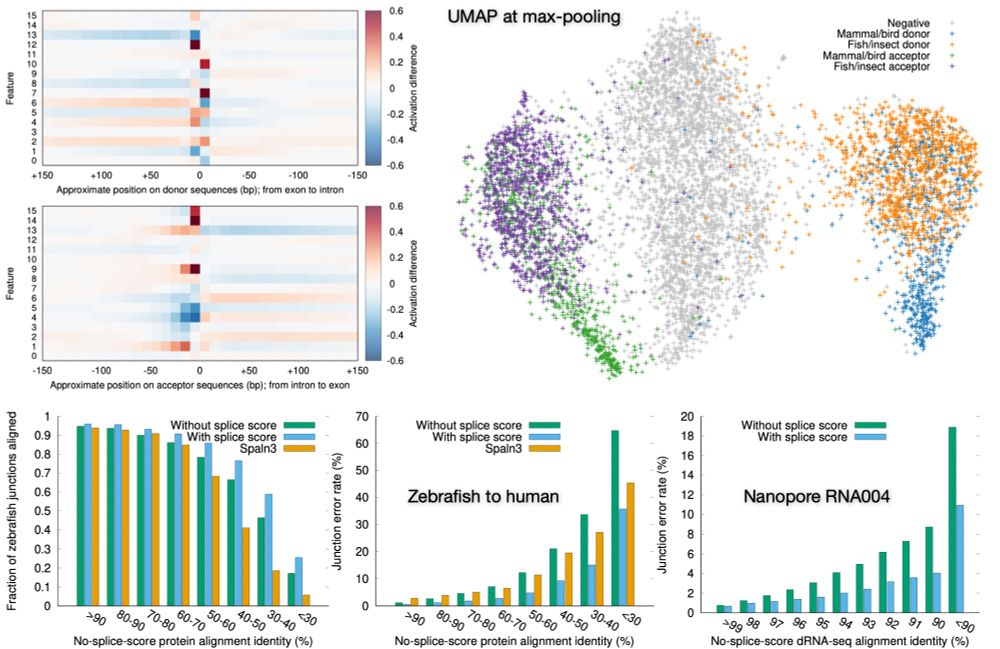
Preprint on "Improving spliced alignment by modeling splice sites with deep learning". It describes minisplice for modeling splice signals. Minimap2 and miniprot now optionally use the predicted scores to improve spliced alignment.
arxiv.org/abs/2506.12986
17.06.2025 01:48 — 👍 109 🔁 54 💬 0 📌 1
Neng Huang developed longcallR for joint SNP calling and phasing from long RNA-seq reads, AND for identifying allele-specific splicing/junctions (ASJ). Although ASJs of statistical significance are rare, a large fraction involve unannotated junctions. In Rust!
30.05.2025 14:54 — 👍 16 🔁 7 💬 0 📌 0
Professor of EECS and Statistics at UC Berkeley. Mathematical and computational biologist.
A microbiologist 🦠& bioinformatician 🧬 uncovering diverse microbial mechanisms via AI ✨. A rising assistant professor @NTU Singapore. With data in the cloud, the sky is the limit! Check out our lab website :) https://genomiverse.net/
Postdoctoral Associate at Yale University
https://medicine.yale.edu/profile/wen-wei-liao
Bioinformatics postdoc at INRAE in Toulouse, France.
Working on data structures and algorithms for pangenome graphs
Bioinformatician/Genomics Software Engineer @garvaninstitute.bsky.social
Views my own.
Mastodon @Psy_Fer_@genomic.social, https://genomic.social
Computational Biologist in Singapore. Nocturnal baker and maker of caffeinated beverages.
Professor, Molecular & Cellular Biology, Harvard University
Bioinformatics Scientist at the Arc Institute.
Working at the intersection of functional genomics, systems biology, and network dynamics. I also build rusty bioinformatics tools
https://github.com/noamteyssier
Bioinformatics. Rust. Mojo.
Genetics & Evolution, Faculty at Stanford & Freeman Hrabowski Scholar at HHMI
schumerlab.com
Bren Professor of Computational Biology @Caltech.edu. Blog at http://liorpachter.wordpress.com. Posts represent my views, not my employer's. #methodsmatter
Associate Professor of CS @ University of Maryland. Proud Rust advocate! I ♥ science & compiled, statically-typed programming languages! Views are my own. Tech stack: https://github.com/rob-p/tech-stack.
Geneticist. Parent. Studying sex chromosomes and sex as a biological variable in disease.
Posts and opinions are my own.
evolutionary genomics of maize and wild relatives. erstwhile ethnobotanist. purveyor of bad puns. dad. prof and HPC director at UC Davis. not actually a cool cow from 1994. rilab.ucdavis.edu. views, especially bad ones, mine only (he/him)
shendure lab |. krishna.gs.washington.edu
Resident panhandler, http://trichelab.org/
Posts may contain trace quantities of blood🩸, chromatin 🧬, and stats 🧮
CV: https://scholar.google.com/citations?user=AOoIO74AAAAJ
Views expressed are my own (but for the right price they can be yours!)
Population and evolutionary genetics @UCDavis. Posts, grammar, & spelling are my views only. He/him. #OA popgen book https://github.com/cooplab/popgen-notes/releases
Executive Director EMBL. I have an insatiable love of biology. Consultant to ONT and Cantata (Dovetail)



![AnniZLab: [🦠, 🧬 , ✨]](https://cdn.bsky.app/img/avatar/plain/did:plc:cz2x2zeakpqqg4qwjytxj25u/bafkreievygd424lvupauxlehnbz6nzzkmsausjscg3adrbdlmax4hdnhce@jpeg)

















