How to Train a Shallow Ensemble
📢 New #preprint is out! Investigating the many flavors of last-layer #UQ, Moritz and 🧑🚀Matthias propose a practitioners' guide on "how to train a shallow ensemble". TL;DR? for good calibration use NLL, include force, and optimize the backbone, fine-tuning for speed! 📃🔗➡️ arxiv.org/html/2602.15...
18.02.2026 16:04 —
👍 3
🔁 2
💬 0
📌 0
ShiftML-3 predictor
No Install. No Setup. Just Chemical Shift Predictions.
At shiftml.materialscloud.io we host the latest ShiftML3 in the web and you can predict chemical shifts of organic crystals for free in the web!
17.02.2026 17:21 —
👍 3
🔁 2
💬 0
📌 0
What a cool applet - running a universal MLIP directly from your webbrowser!
30.11.2025 19:12 —
👍 0
🔁 0
💬 0
📌 0

error plots for the PET-MAD-DOS model on different datasets
Anticipating 🧑🚀 Wei Bin's talk at #psik2025 (noon@roomA), 📢 a new #preprint using PET and the MAD dataset to train a universal #ml model for the density of states, giving band gaps for solids, clusters, surfaces and molecules with MAE ~200meV. Go to the talk, or check out arxiv.org/html/2508.17...!
28.08.2025 07:19 —
👍 4
🔁 2
💬 0
📌 0
First, we generate a pool of candidate structures and then select the one whose calculated chemical shieldings best match the experimental measurements. Because shielding calculations quickly become the computational bottleneck, machine-learning (ML) models can substantially reduce the cost.
25.08.2025 08:53 —
👍 0
🔁 0
💬 1
📌 0
Solid-state NMR spectroscopy is increasingly used for structure determination of organic solids. NMR-based structure determination differs from conventional diffraction methods:
25.08.2025 08:53 —
👍 1
🔁 0
💬 1
📌 0
GitHub - lab-cosmo/shiftml: A python package for the prediction of chemical shieldings of organic solids and beyond.
A python package for the prediction of chemical shieldings of organic solids and beyond. - lab-cosmo/shiftml
We're introducing ShiftML3, a new ShiftML model for chemical shielding predictions in organic solids.
* ShiftML3 predicts full chemical shielding tensors
* DFT accuracy for 1H, 13C, and 15N
* ASE integration
* GPU integration
Code: github.com/lab-cosmo/Sh...
Install from Pypi: pip install shiftml
25.08.2025 08:53 —
👍 2
🔁 3
💬 1
📌 0

metatensor logo

metatomic logo
🚨 #machinelearning for #compchem goodies from our 🧑🚀 team incoming! After years of work it's time to share. Go check arxiv.org/abs/2508.15704 and/or metatensor.org to learn about #metatensor and #metatomic. What they are, what they do, why you should use them for all of your atomistic ML projects 🔍.
22.08.2025 07:40 —
👍 12
🔁 8
💬 1
📌 2
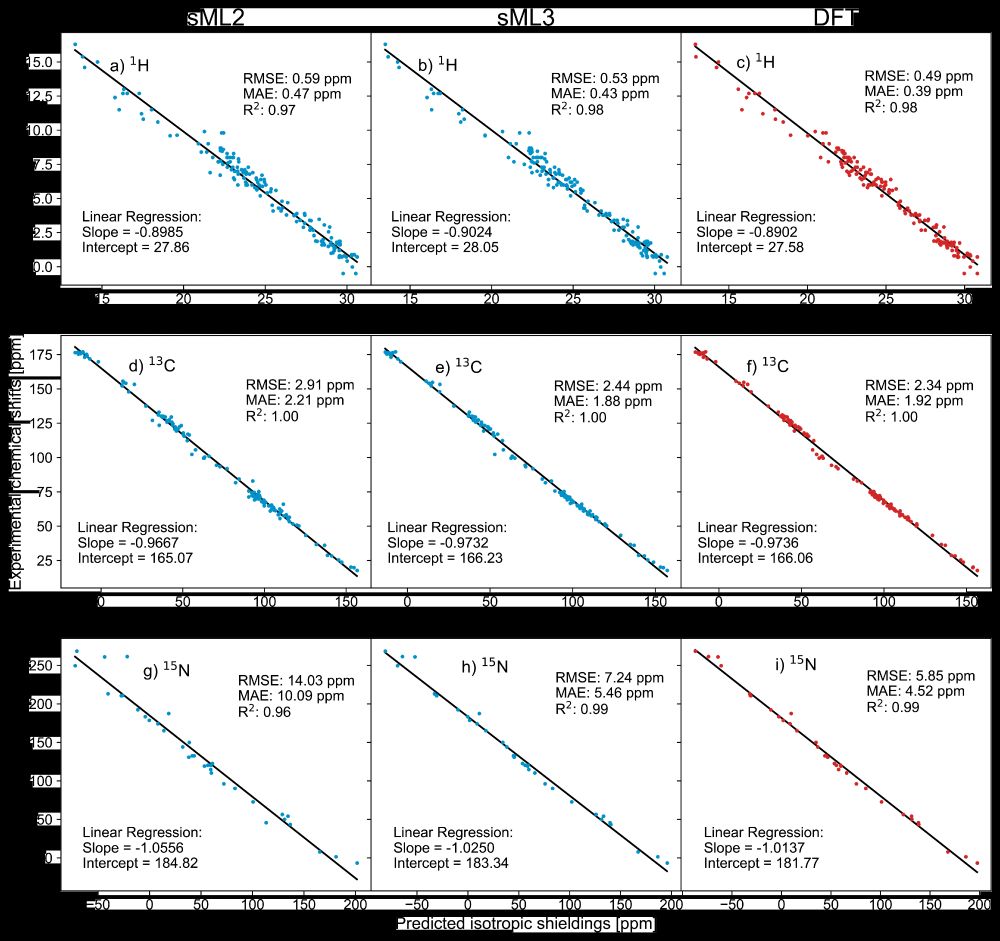
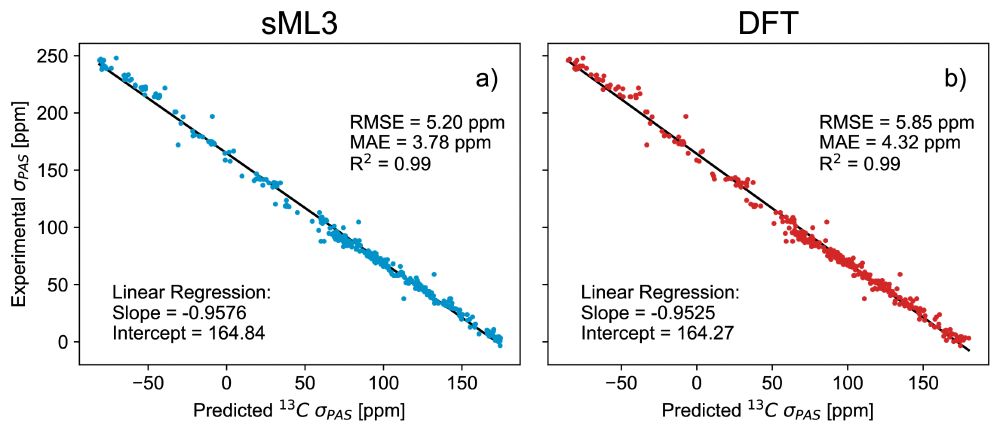
🎉 DFT-accurate, with built-in uncertainty quantification, providing chemical shielding anisotropy - ShiftML3.0 has it all! Building on a successful @nccr-marvel.bsky.social-funded collaboration with LRM🧲⚛️, it just landed on the arXiv arxiv.org/html/2506.13... and on pypi pypi.org/project/shif...
17.06.2025 13:18 —
👍 18
🔁 7
💬 1
📌 0
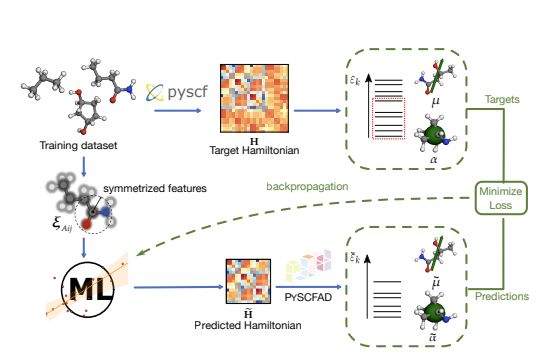
A schematic of the functioning of a ML/QM hybrid framework
When you combine #machinelearning and #compchem, you need to start worrying at the QM details within your ML architecture. We use our indirect Hamiltonian framework and pySCFAD to explore the enormous design space arxiv.org/abs/2504.01187
03.04.2025 21:30 —
👍 14
🔁 4
💬 2
📌 0
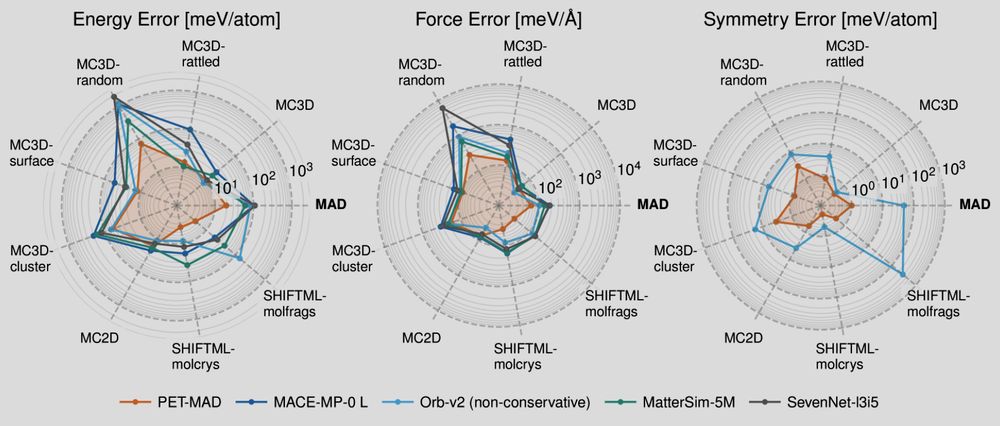
Polar plot showing the errors of several machine-learning potential of different test sets. Smaller is better here!
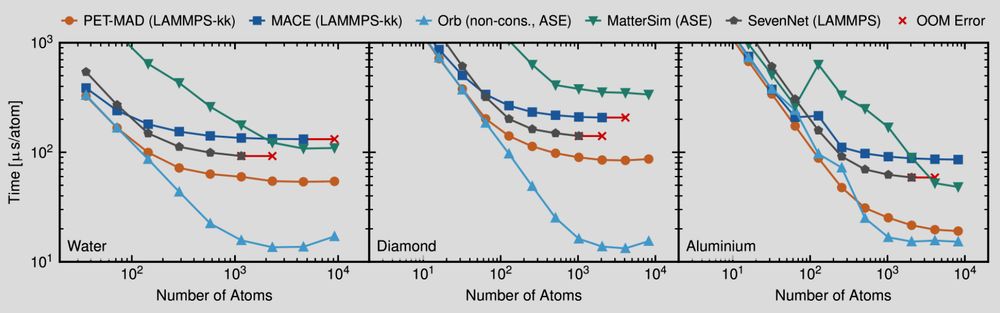
Plots showing the evaluation time per atom for several machine-learning potentials as a function of the number of atoms in a simulation. Smaller is better
📢 PET-MAD has just landed! 📢 What if I told you that you can match & improve the accuracy of other "universal" #machinelearning potentials training on fewer than 100k atomic structures? And be *faster* with an unconstrained architecture that is conservative with tiny symmetry breaking? Sounds like 🧑🚀
19.03.2025 07:23 —
👍 28
🔁 9
💬 1
📌 3

Header of the webpage showing the title ("Atomistic Water model for MD") and the authors (Philip Loche, Marcel Langer, Michele Ceriotti)
Happy to share a new #cookbook recipe that shocases several new software developments in the lab, using the good ole' QTIP4P/f water model as an example. atomistic-cookbook.org/examples/wat.... TL;DR - you can now build torch-based interatomic potentials, export them and use them wherever you like!
28.02.2025 12:58 —
👍 12
🔁 5
💬 1
📌 0
Feeling a bit lonely here ...
15.11.2024 11:38 —
👍 0
🔁 0
💬 0
📌 0


