
New tool from @alexsweeten.bsky.social to find and classify all your satellites: "AniAnn's: alignment-free annotation of tandem repeat arrays using fast average nucleotide identity estimates"
📄 www.biorxiv.org/content/10.6...
📦 github.com/marbl/anianns
29.01.2026 13:05 — 👍 44 🔁 19 💬 4 📌 1
HLi Lab - Vacancies
Openings
I am looking for a postdoc to develop high-performance algorithms in computational genomics. Email or DM me if interested. For more information, see hlilab.github.io/vacancies. RTs appreciated!
14.01.2026 15:44 — 👍 43 🔁 64 💬 1 📌 0
(t)rust the process?
23.11.2025 06:35 — 👍 1 🔁 0 💬 0 📌 0
Really excited to see our new work in scaling Mumemto to any size pangenome published in Genome Research this morning. And right on cue with the great opportunity to present this work at #GI2025 this week.
07.11.2025 21:29 — 👍 16 🔁 5 💬 1 📌 0

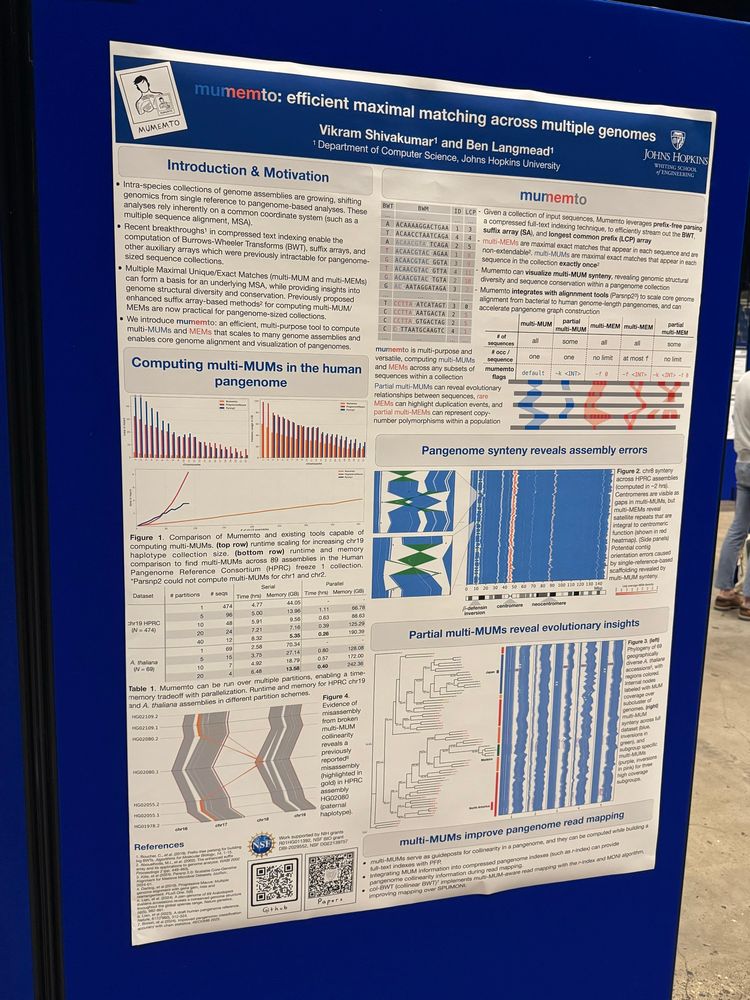
Figure 1: (A) Anchor-based merging requires a common sequence (red) present in each partition. Multi-MUMs are merged by identifying overlaps between partition-specific matches in the anchor coordinate space, and a uniqueness threshold determines if a MUM is still unique in each partition after truncation. (B) String-based merging enables compu- tation of multi-MUMs between partitions without a common sequence. An example tree (left) is shown, highlighting the use case where partial multi-MUMs specific to internal nodes (starred) can be computed by merging subclade-based partitions up a tree. (right) MUM overlaps are computed by running Mumemto on the MUM sequences, and the uniqueness threshold array ensures overlaps remain unique across the merged dataset. (C) An example Burrows-Wheeler Transform (BWT), matrix (BWM), and Longest Com- mon Prefix (LCP) array, with sequence IDs for each suffix shown (ID). A non-maximal unique match (UM) is shown, and the uniqueness threshold for this match is found us- ing the flanking LCP values. (D) A partial multi-MUM (in blue) is found in all-but-one sequence (excluded in red). Using two anchor sequences (red and orange), all-but-one partial MUMs can be computed using an augmented anchor-based merging method (sec- tion 2.6).


Fantastic talk by @vikramshivakumar.bsky.social Mumemto—Scalable multi-MUM finding for pangenomes
Papers biorxiv.org/content/10.1101/2025.05.20.654611 & doi.org/10.1186/s13059-025-03644-0
Code: github.com/vikshiv/mume...
Very efficient pangenome visualization tool, revealing synteny and variations!
06.11.2025 01:13 — 👍 23 🔁 12 💬 1 📌 1
Looking forward to lots of great talks from JHU folks at CSHL this week!
04.11.2025 18:20 — 👍 4 🔁 0 💬 0 📌 0
Now this, undergrads, is how you cold email a professor.
03.11.2025 20:32 — 👍 2 🔁 0 💬 0 📌 0

If that’s not enough, we threw in a complete, T2T giraffe genome! Giraffe genomes are pretty cool. Almost all of their chromosomes are Robertsonian fusions of the typically telocentric ruminant chromosomes. 🐄 vs. 🦒...
10.10.2025 15:26 — 👍 9 🔁 4 💬 2 📌 0
Last week we were in the Washington Post for our characterization of Robertsonian chromosomes. This week we are entering our 10th day of being shut down and all of our research is on hold. To help me feel not-so-bad, here is a thread of some studies we released right before the shutdown 🧵 [1/n]...
10.10.2025 15:24 — 👍 48 🔁 17 💬 1 📌 0

Figure 1(A) ANI quantifies the similarity between two genomes. ANI can be defined as the number of aligned positions where the two aligned bases are identical, divided by the total number of aligned bases. Historically, ANI was calculated using a single gene family for multiple sequence alignment. Another approach finds orthologous genes between two genomes and reports the average similarity between their CDSs. This method was later extended to whole-genome alignment by identifying local alignments and excluding supplementary alignments with lower similarity. (B) Different ANI tools employ various approaches in calculating ANI values. ANIm, OrthoANI, and FastANI use aligners to identify homologous regions, whereas Mash uses k-mer hashing to estimate similarities. Only alignments with higher similarity represented by green arrows are included in ANI calculations, while red arrows, corresponding to paralogs, are excluded. (C) The proposed benchmarking method evaluates the performance of different tools using both real and simulated data. It assumes that more distantly related species on the phylogenetic tree should have lower ANI similarities. This is measured by calculating the statistics of Spearman rank correlation. We expect a negative correlation between ANI and the tree distance (scatter plot on the right).
https://academic.oup.com/bib/article/doi/10.1093/bib/bbaf267/8160681
Excited to share our EvANI benchmarking workflow, published in Briefings in Bioinformatics doi.org/10.1093/bib/...
Computing average nucleotide identity (ANI) is neither conceptually nor computationally trivial. Its definition has evolved over years, with different meanings and assumptions (1/5)
21.09.2025 15:26 — 👍 30 🔁 12 💬 1 📌 1
10/10 tool name 👌
22.08.2025 13:43 — 👍 3 🔁 0 💬 1 📌 0

Anchor-based merging requires a common sequence (red) present in each partition. Multi-MUMs are merged by identifying overlaps between partition-specific matches in the anchor coordinate space, and a uniqueness threshold determines if a MUM is still unique in each partition after truncation. (B) String-based merging enables computation of multi-MUMs between partitions without a common sequence. An example tree (left) is shown, highlighting the use case where partial multi-MUMs specific to internal nodes (starred) can be computed by merging subclade- based partitions up a tree. (right) MUM overlaps are computed by running Mumemto on the MUM sequences, and the uniqueness threshold array ensures overlaps remain unique across the merged dataset. (C) An example Burrows-Wheeler Transform (BWT), matrix (BWM), and Longest Common Prefix (LCP) array, with sequence IDs for each suffix shown (ID). A non-maximal unique match (UM) is shown, and the uniqueness threshold for this match is found using the flanking LCP values. (D) A partial multi-MUM (in blue) is found in all-but-one sequence (excluded in red). Using two anchor sequences (red and orange), all-but-one partial MUMs can be computed using an augmented anchor-based merging method.


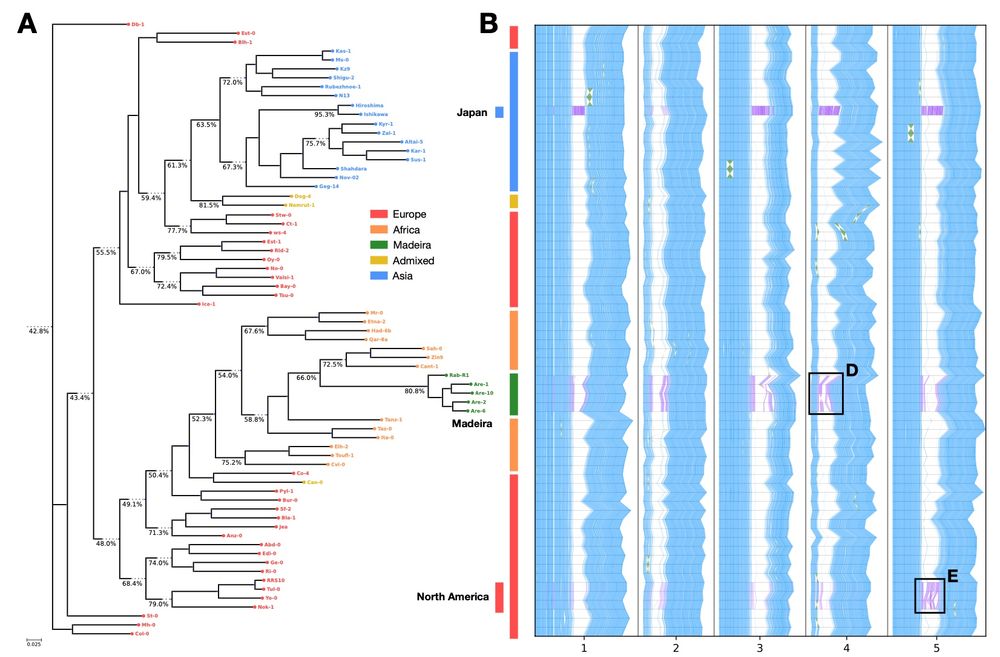
(A) Phylogeny of geographically diverse A. thaliana accessions (Lian et al. 2024), with broad geographical regions colored. Internal nodes are labeled with the coverage of partial multi-MUMs across the leaves of each node. Internal node partial MUMs are computed by merging subtree-based partitions progressively up the phylogeny. (B) Global multi-MUM synteny across the full dataset shown in blue (with inversions in green). Global MUMs are computed by merging all partitions together (representing the root node). Additionally, three geographically distinct subgroups are highlighted and partition-specific multi-MUMs (in purple, with inversions in pink) reveal local structural variation in centromeric regions.
Great talk by Vikram @vikramshivakumar.bsky.social on studying pangenomes and synteny visualization in #WABI25
Github: github.com/vikshiv/mume...
First paper: genomebiology.biomedcentral.com/articles/10....
Second: www.biorxiv.org/content/10.1... #WABI2025
20.08.2025 15:03 — 👍 22 🔁 8 💬 0 📌 0

Vikram Shivakumar telling us about "Partitioned Multi-MUM finding for scalable pangenomics" #WABI25! So many kinds of matches!
20.08.2025 14:33 — 👍 8 🔁 3 💬 0 📌 0
Not saying I agree either way, but one pro for text-based file formats are less dependencies needed for viewing files
06.08.2025 22:42 — 👍 0 🔁 0 💬 1 📌 0
This is so amazing, thank you!
31.07.2025 13:00 — 👍 1 🔁 0 💬 0 📌 0

comic doodle of Vikram Shivakumar in a sweater and checkered shirt on a pink gradient background, with various elements of the talk to the left: two old moms pointing at MUMs, below an explanation of what those are (large chunks of the same DNA sequence through the genome), at the bottom a few of the organisms worked on: a tomato, a potato, an arabidopsis weed.
#SciArt doodle of @vikramshivakumar.bsky.social's talk yesterday at the @sangerinstitute.bsky.social on MUMs*
*maximal unique matches in pangenomes, now if you did that on sequenced moms you could do mummoms
31.07.2025 07:51 — 👍 18 🔁 2 💬 2 📌 0
Excited to share our new preprint on detecting foldback artifacts in long reads with my advisors Matthew Meyerson and @lh3lh3.bsky.social ! Stop by poster C-180 on Wednesday at ISMB/ECCB2025 to learn more and chat!
21.07.2025 14:26 — 👍 4 🔁 2 💬 0 📌 0
And of course, the poster itself:
21.07.2025 17:00 — 👍 3 🔁 0 💬 0 📌 0
If you’re in Liverpool, stop by my poster A217 at ISMB/EECB 2025, and chat about all things pangenomes, MUMs, and alignment (and the Beatles or Oasis-mania)
21.07.2025 15:11 — 👍 16 🔁 6 💬 2 📌 0
Really excited to see this published! To more mum-finding 🍻
17.06.2025 15:01 — 👍 21 🔁 7 💬 2 📌 0
We are what we index; a primer for the Wheeler Graph era
Talk by Ben Langmead - WABI 2025
🖥️🧬We're thrilled to announce that one of our keynote speakers at #WABI2025 will be the inimitable @benlangmead.bsky.social! wabiconf.github.io/2025/talks/t... Ben's keynote is titled "We are what we index; a primer for the Wheeler Graph era", & it's sure to be a whirlwind tour of full-text indexing!
16.06.2025 12:46 — 👍 20 🔁 5 💬 1 📌 0
1/5 We introduce Movi Color, led by Steven Tan (a brilliant undergrad member of Langmead lab) for taxonomic and multi-class classification. It uses a full-text index based on the move structure and does not rely on predefined values (like k-mer length) for index building.
github.com/mohsenzakeri...
29.05.2025 14:36 — 👍 15 🔁 6 💬 1 📌 2
GitHub - vikshiv/mumemto: Mumemto: multi-MUM and MEM finding across pangenomes
Mumemto: multi-MUM and MEM finding across pangenomes - vikshiv/mumemto
We've released a new version (v1.3) of Mumemto (github.com/vikshiv/mume...) that implements merging. Running Mumemto in merge-mode makes the output set of multi-MUMs dynamic, so adding new assemblies is as easy as computing a new set of MUMs and merging them in.
27.05.2025 19:35 — 👍 5 🔁 0 💬 0 📌 0
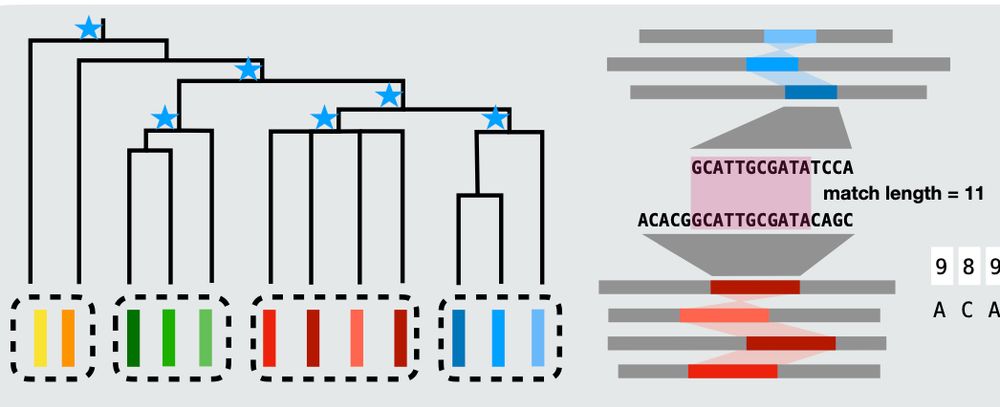
We can also merge along the shape of a phylogenetic tree, finding clade-specific variation and conserved elements. Previously, adding new assemblies can lose MUMs, which must be present across the whole collection. Now we can find MUMs that reveal local variation distinct to specific subgroups. 3/n
27.05.2025 19:35 — 👍 4 🔁 2 💬 1 📌 0
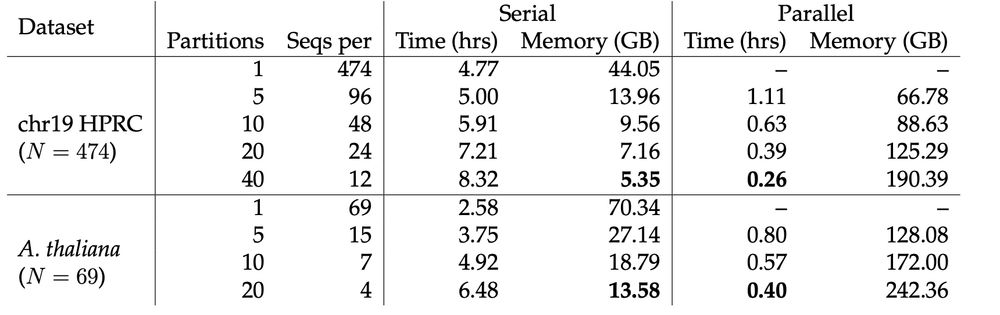
We implement two partition/merge algorithms that can merge multi-MUMs between datasets. This makes Mumemto highly parallelizable, but also very memory efficient if partitions are computed in serial. 2/n
27.05.2025 19:35 — 👍 2 🔁 0 💬 1 📌 0

Partitioned Multi-MUM finding for scalable pangenomics
Pangenome collections are growing to hundreds of high-quality genomes. This necessitates scalable methods for constructing pangenome alignments that can incorporate newly-sequenced assemblies. We prev...
Excited to share a new update to Mumemto, scaling MUM and conserved element finding to any size pangenome! Preprint out now w/ @benlangmead.bsky.social.
Mumemto scales to the new HPRC v2 release and beyond, and can merge in future assemblies without any recomputation! 1/n
27.05.2025 19:35 — 👍 27 🔁 15 💬 1 📌 2
Our pre-print on investigating variation in South Asian genomes is now out!
Thank you to @mikeschatz.bsky.social, @rajivmccoy.bsky.social and @aabiddanda.bsky.social for all their work on this.
🧵 A thread on the key results and takeaways from our work:
15.05.2025 14:19 — 👍 23 🔁 9 💬 2 📌 6
Graduate Student Researcher,
Computational Genomics Lab,
UC Santa Cruz Genomics Institute.
Sudmantlab.org - Assistant professor Berkeley
Postdoctoral Associate at Yale University
https://medicine.yale.edu/profile/wen-wei-liao
Computational and evolutionary genetics professor at the University of Cambridge, and Associate Faculty at the Wellcome Sanger Institute. Open access genomes, at scale, for all of life.
Rice CS PhD Student | Treangen Lab + Tisza Lab (BCM) | Bioinformatics and Viromics | Former CWRU Men's Soccer
Associate Professor and Research Dean at UVA School of Data Science, #Rstats enthusiast, dad, runner, guitar noise-maker. Views my own.
Web: https://datascience.virginia.edu/people/stephen-turner
Newsletter: https://blog.stephenturner.us
CS + Applied Math @ JHU | Undergrad @ Battle Lab | Mountain Enthusiast
PhD student @ Ca' Foscari University of Venice
PhD student at EMBL-EBI and the University of Cambridge
Finn @robdfinn.bsky.social and Lees Group @bacpop.org
Bioinformatics Scientist at the Arc Institute.
Working at the intersection of functional genomics, systems biology, and machine learning. I also build rusty bioinformatics tools
https://github.com/noamteyssier
Human evolutionary genetics, University of Cambridge; Darwin College. 🇮🇪
PhD student in the Schatz lab @ Johns Hopkins University
Assistant professor @TGen | Postdoc @jacksonlab @MDAndersonNews | Genomics | Cancer | Gliomas | Telomeres
Bioinformatics Scientist at Illumina. I work on finding challenging variants. Views are my own.
https://orcid.org/0000-0002-8605-7547
PhD student in the Lees group @ Cambridge Uni & EMBL-EBI | bioinformatics | pathogen genomics | Rust, Stan, python, R
Assistant Professor at JHU CS developing statistical/ML methods for biological applications
Associate Professor at Johns Hopkins studying human evolutionary and reproductive genetics https://mccoy-lab.org
Scientist at NHGRI/NIH. Posts are my opinions only. She/her
























