
1/6 Movi 2 is here: faster and more space-efficient for pangenome queries. Its fastest mode uses half the memory of Movi 1 while running ~30% faster. github.com/mohsenzakeri...
21.10.2025 20:00 — 👍 44 🔁 24 💬 1 📌 2
@danydoerr.bsky.social
1/6 Movi 2 is here: faster and more space-efficient for pangenome queries. Its fastest mode uses half the memory of Movi 1 while running ~30% faster. github.com/mohsenzakeri...
21.10.2025 20:00 — 👍 44 🔁 24 💬 1 📌 2
Wie gesund ist eigentlich Eisbaden?
Patrick Schrauwen vom @ddzdiabetes.bsky.social - Leibniz-Zentrum für Diabetes-Forschung an der @hhu.de in der neuen Folge von #TonspurWissen, dem #Podcast von Rheinischer Post und @leibniz-gemeinschaft.de.
👉 pod.fo/e/33e7e0 ❄️
#Gesundheit #Diabetes #Eisbaden

I am hiring! - looking for a Staff Scientist to co-run my research group with me. Staff Scientist is a senior professional scientist role at EMBL. Please forward to people you might know who could be interested! embl.wd103.myworkdayjobs.com/en-US/EMBL/j...
10.10.2025 07:30 — 👍 108 🔁 118 💬 2 📌 5
Have you recently completed (or finishing soon) a PhD in CS or a related discipline? Do you want to do research advancing the theory & practice of algorithmic genomics & build tools that people love to use? I'll be looking to hire a postdoc! Official ad coming soon:
docs.google.com/document/d/1...

Vikram Shivakumar telling us about "Partitioned Multi-MUM finding for scalable pangenomics" #WABI25! So many kinds of matches!
20.08.2025 14:33 — 👍 8 🔁 3 💬 0 📌 0
"Human readable compression of GFA paths using grammar-based code" being presented by Peter Heringer at #WABI25
20.08.2025 17:42 — 👍 9 🔁 3 💬 0 📌 0
Anchor-based merging requires a common sequence (red) present in each partition. Multi-MUMs are merged by identifying overlaps between partition-specific matches in the anchor coordinate space, and a uniqueness threshold determines if a MUM is still unique in each partition after truncation. (B) String-based merging enables computation of multi-MUMs between partitions without a common sequence. An example tree (left) is shown, highlighting the use case where partial multi-MUMs specific to internal nodes (starred) can be computed by merging subclade- based partitions up a tree. (right) MUM overlaps are computed by running Mumemto on the MUM sequences, and the uniqueness threshold array ensures overlaps remain unique across the merged dataset. (C) An example Burrows-Wheeler Transform (BWT), matrix (BWM), and Longest Common Prefix (LCP) array, with sequence IDs for each suffix shown (ID). A non-maximal unique match (UM) is shown, and the uniqueness threshold for this match is found using the flanking LCP values. (D) A partial multi-MUM (in blue) is found in all-but-one sequence (excluded in red). Using two anchor sequences (red and orange), all-but-one partial MUMs can be computed using an augmented anchor-based merging method.


(A) Phylogeny of geographically diverse A. thaliana accessions (Lian et al. 2024), with broad geographical regions colored. Internal nodes are labeled with the coverage of partial multi-MUMs across the leaves of each node. Internal node partial MUMs are computed by merging subtree-based partitions progressively up the phylogeny. (B) Global multi-MUM synteny across the full dataset shown in blue (with inversions in green). Global MUMs are computed by merging all partitions together (representing the root node). Additionally, three geographically distinct subgroups are highlighted and partition-specific multi-MUMs (in purple, with inversions in pink) reveal local structural variation in centromeric regions.
Great talk by Vikram @vikramshivakumar.bsky.social on studying pangenomes and synteny visualization in #WABI25
Github: github.com/vikshiv/mume...
First paper: genomebiology.biomedcentral.com/articles/10....
Second: www.biorxiv.org/content/10.1... #WABI2025
The 25th iteration of the excellent Conference for Algorithms in Bioinformatics (WABI) starts tomorrow at UMD @umdscience.bsky.social at the Brendan Iribe Center. You can find details at the website wabiconf.github.io/2025/. We'll use the tag #WABI25 for the meeting!
20.08.2025 02:43 — 👍 17 🔁 9 💬 0 📌 0
We open our second day at #Hitseq @hitseq.bsky.social with our last key note speaker Tobias Marschall and his insightful talk about the human pangenome and the challenges for structural variation analysis. His work has focused on tackling current limitations such as sample size and remaining gaps
24.07.2025 09:01 — 👍 8 🔁 3 💬 0 📌 0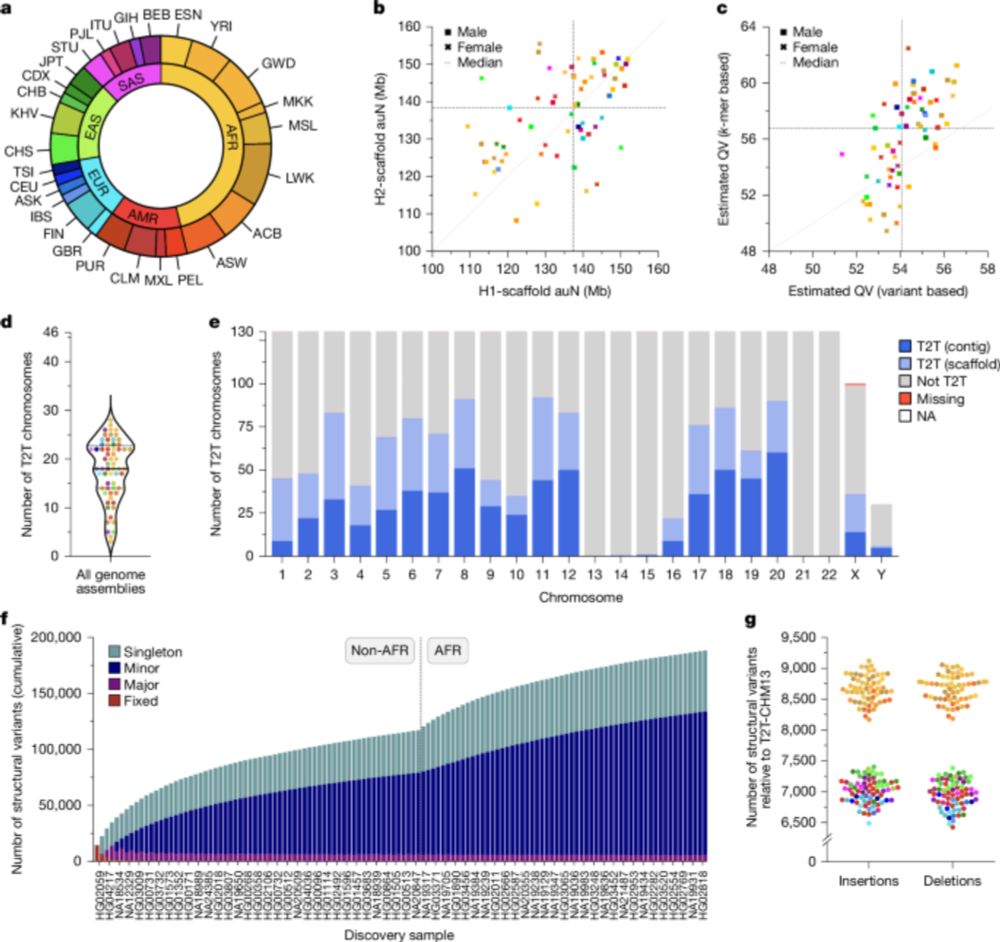
Two papers in today's issue of @nature.com : 1) we assemble 65 genomes to near completion, including centromeres and the MHC. tinyurl.com/3huhax6w. 2) we sequence 1,019 genomes from the 1kGP with long reads, revealing SVs down to low allele frequencies tinyurl.com/wbx3we9x.
23.07.2025 15:12 — 👍 55 🔁 24 💬 1 📌 2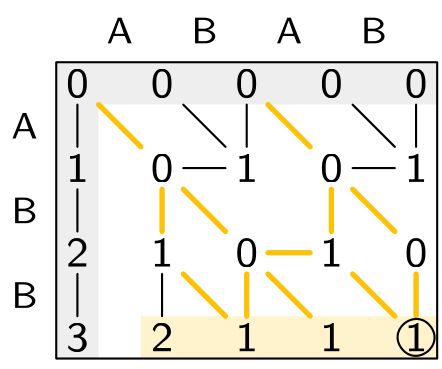
Sassy solves approximate string matching: finding all matches of a pattern in a text.
Sassy is out now!
Ever need to search for approximate matches of short DNA strings?
Sassy is the tool to use!
Available now wherever you get your code
With @rickbitloo.bsky.social
curiouscoding.nl/papers/sassy...
github.com/ragnarGrootK...

@denbi.bsky.social will offer 3 #workshops at @gcb-bioinformatics.bsky.social 2025, focusing on #cloudcomputing, #pangenomics and #protein structure analysis. For more information see: www.denbi.de/news1/1904-d...
12.06.2025 09:04 — 👍 5 🔁 4 💬 0 📌 0
🧬🖥️
📝 To-Do List for #GCB2025:
✅ Check the programme: gcb2025.de
✅ Submit your poster
✅ Buy your conference ticket
✅ Book your hotel room
✅ Take advantage of the DB event offer and travel to Düsseldorf by train at a discounted rate, more info here: gcb2025.de/GCB2025_venu...
Now published! Note that since Vikram's original post (quoted here), he's made it easy to dynamically update a set of multi-MUMs (e.g. when more genomes are added to a pangenome) and to find multi-MUMs for huge collections like HPRCv2 genomebiology.biomedcentral.com/articles/10....
17.06.2025 14:02 — 👍 53 🔁 22 💬 1 📌 1🆕 New Features
• Re-rendering of HTML
• Plug in your own reports with !Custom
• Download plots as PNG or SVG
• Select hexagons in NodeDistribution 🔷 and download node ids!
🐞 Fixes & Improvements
• 🐌 Faster similarity calculation
• 🔧 Cleaner YAML syntax
• 🔁 Better handling of dependent analyses

🚀 Panacus v0.4.0 is out!
Bringing more power, flexibility, and speed to your pangenome reports 🧬📊
👉 Upgrade now & explore: github.com/codialab/pan...
#genomics #bioinformatics #pangenome #panacus
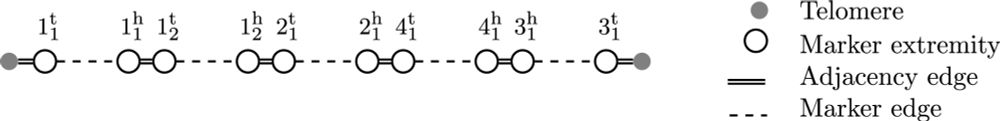
Improved method for inferring ancestral genomes! We introduce a powerful ILP for the Small Parsimony Problem under a complex rearrangement model that handles duplications & indels. Big gains in accuracy & runtime on real and simulated data! #Genomics #Phylogenetics
doi.org/10.1186/s130...
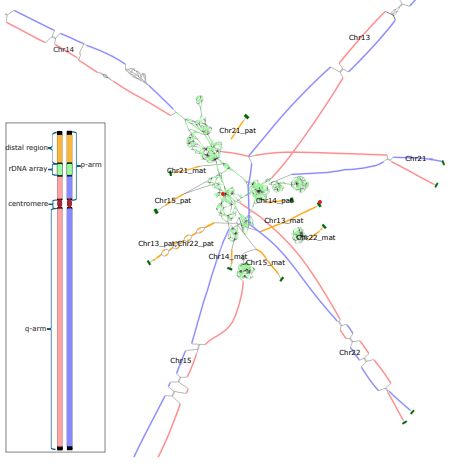
Verkko2 integrates proximity ligation data with long-read De Bruijn graphs for efficient telomere-to-telomere genome assembly, phasing, and scaffolding. #LongReads #HiC #GenomeAssembly #GenomeScaffolding #Bioinformatics @genomeresearch.bsky.social
genome.cshlp.org/content/earl...

🧬 & 🖥️
📢Call for Posters #GCB2025 | Join the #bioinformatics and #computationalbiology community at the German Conference on Bioinformatics! Submit your poster now!
Important Deadlines:
📌 Early Bird Registration: 5 July 25
📌 Poster Submission: 7 August 25
🔗 gcb2025.de/GCB2025_call...
Slides from my talk (with @kamilsjaron.bsky.social) on an history of k-mers in bioinformatics: rayan.chikhi.name/pdf/2025-kme...
03.06.2025 09:25 — 👍 44 🔁 24 💬 1 📌 2
New in pangenomics: We extend the GFA format to represent haplotype paths in a compressed yet human-readable way. Our tool sqz (/ˈskuzi/) achieves 40× compression on chr19 with 1000 haplotypes, plus 10× faster analysis and 8× lower memory use.
🔗 www.biorxiv.org/content/10.1...
#bioinformatics

Submit your abstracts from all areas of #bioinformatics and #computationalbiology for the German Conference on Bioinformatics - #GCB2025 at University of Dusseldorf from 22 - 24 September 2025!
abstract submission: gcb2025.de
@denbi.bsky.social @openbio.bsky.social @SIB.mstdn.science.ap.brid.gy


















