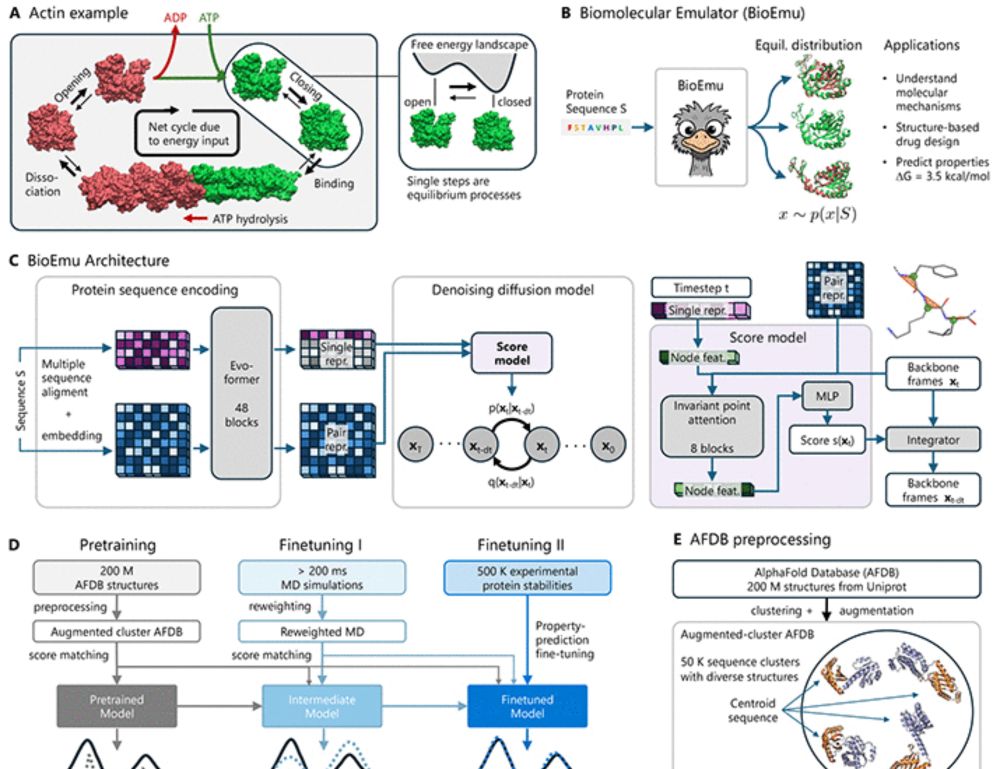
Preprint release 😀 of "Speak to a Protein," an AI co-scientist that facilitates data gathering and analysis in an interactive collaborative session. It is quite amazing to use. Preprint: arxiv.org/abs/2510.17826
22.10.2025 16:39 — 👍 8 🔁 4 💬 0 📌 0

Crystallography for the "dark proteome"? From @nudrugdiscovery.bsky.social and @chemistryncl.bsky.social : #FragLites map protein–protein interaction sites including regions with no previously known function. 🧵1/3 #DrugDiscovery #ChemBio
📖 www.sciencedirect.com/science/arti...
14.08.2025 15:33 — 👍 15 🔁 6 💬 1 📌 0

"Enhancing Electrostatic Embedding for ML/MM Free Energy Calculations", by Joao Morado et al, is now available on ChemRxiv! doi.org/10.26434/che... #compchem
23.06.2025 11:52 — 👍 11 🔁 3 💬 2 📌 0
EPSRC Postdoctoral Pathway Fellowship
EPSRC Postdoctoral Pathway Fellowship
Are you an EPSRC funded PGR? Ready to submit your thesis this year, or just passed your viva and looking for a postdoc position? Get in touch if interested in apply for a pathway fellowship in the areas of molecular modelling or computer-aided drug design ⬇️
jobs.ncl.ac.uk/job/Newcastl...
13.06.2025 15:55 — 👍 2 🔁 1 💬 0 📌 0

📢 New preprint: "A graph neural network charge model targeting accurate electrostatic properties of organic molecules" by @charlie-adams.bsky.social et al out now on @chemrxiv.bsky.social #compchem
doi.org/10.26434/che...
03.06.2025 14:47 — 👍 13 🔁 3 💬 1 📌 0

The CCPBioSim Annual Conference - Frontiers in Biomolecular Simulations will be taking place in Southampton, 14-16 July 2025. Registration is now open! Details and the registration link can be found at www.ccpbiosim.ac.uk/soton2025 #compchem
10.05.2025 12:42 — 👍 5 🔁 4 💬 0 📌 0
Postdoctoral Fellowships
The information provided on this page is a summary of the main rules and requirements for Postdoctoral Fellowships (PFs) and who can apply for them.
Passionate about force fields? Got great ideas for the future of force field design?
We are looking to support applicants to the #MSCA Postdoctoral Fellowships scheme in collaboration with @openforcefield.org!
The call opens soon, get in touch if interested ⬇️ tinyurl.com/yxbpj4y4
22.04.2025 15:21 — 👍 6 🔁 4 💬 1 📌 1
Visualizations in the OpenFF Toolkit — OpenFF Ecosystem documentation
The OpenFF toolkit contains some facilities for visualizing molecules in a Jupyter notebook, which can be a convenient way to inspect the system you're setting up.
docs.openforcefield.org/en/latest/ex...
#compchem
Share your own examples of visualizing molecules!
18.03.2025 15:56 — 👍 5 🔁 2 💬 0 📌 0

Header of the webpage showing the title ("Atomistic Water model for MD") and the authors (Philip Loche, Marcel Langer, Michele Ceriotti)
Happy to share a new #cookbook recipe that shocases several new software developments in the lab, using the good ole' QTIP4P/f water model as an example. atomistic-cookbook.org/examples/wat.... TL;DR - you can now build torch-based interatomic potentials, export them and use them wherever you like!
28.02.2025 12:58 — 👍 12 🔁 5 💬 1 📌 0
Excited to share what I’ve been working on with @tkaraletsos.bsky.social to transform the field of drug discovery with atomistic foundation simulation models!
I could not be more thrilled to be working with this dream team.
achira.ai
21.02.2025 14:15 — 👍 90 🔁 17 💬 9 📌 2

CCPBioSim Annual Conference 2025
More detail will be added here very soon!
📢 SAVE THE DATE: This year's annual CCPBioSim conference will be in sunny Southampton on the 14th-16th July 2025.
Stay tuned on here and at ccpbiosim.ac.uk/soton2025 for more information and announcements coming soon!
#compchem #chemsky
20.02.2025 16:31 — 👍 6 🔁 3 💬 0 📌 0
SAVE THE DATE: We’re hosting this years annual CCPBioSim conference in sunny Southampton on the 14th-16th July 2025. Stay tuned on socials and at ccpbiosim.ac.uk/soton2025 for more information and announcements coming soon!
17.02.2025 16:41 — 👍 1 🔁 1 💬 0 📌 1
Thanks to Julien Michel and @colegroupncl.bsky.social
for supervision. Please try the methods and let us know how they work on your data! Feedback is welcome.
(6/6)
24.01.2025 11:20 — 👍 2 🔁 0 💬 0 📌 0

GitHub - fjclark/red: Robust Equilibration Detection
Robust Equilibration Detection. Contribute to fjclark/red development by creating an account on GitHub.
We implement all methods in the Python package RED ("conda install conda-forge::red-molsim", github.com/fjclark/red) and provide a complete workflow to reproduce the work (github.com/michellab/Ro...). All data are available on Zenodo (zenodo.org/records/1390...).
(5/6)
24.01.2025 11:20 — 👍 4 🔁 0 💬 1 📌 0

We observe a general trade-off: methods which more thoroughly account for autocorrelation often discard too much data, while methods which less thoroughly account for autocorrelation often discard too little data. We recommend a method which balances these extremes.
(4/6)
24.01.2025 11:20 — 👍 3 🔁 0 💬 1 📌 0

To quantitatively assess the heuristics, we create sets of synthetic data modelled on long alchemical absolute binding free energy calculations. Since we know the true unbiased mean of our synthetic data, we can calculate the errors each of the heuristics introduces.
(3/6)
24.01.2025 11:20 — 👍 4 🔁 0 💬 1 📌 0

We test a range of truncation point selection heuristics. Following White (doi.org/10.1177/0037...), these all work by minimising the marginal standard error, but differ in how they account for autocorrelation.
(2/6)
24.01.2025 11:20 — 👍 4 🔁 0 💬 1 📌 0
PhD student with @compbiophys | Toronto 🇨🇦 x Göttingen 🇩🇪
PhD student at MPINAT. Microtubules and coarse grained simulations.
👨💻PhD candidate
💊Structure-based & Computational Drug Discovery
📍Newcastle University, Newcastle upon Tyne, UK
Theoretical Chemist; Group leader at the Wrocław University of Science and Technology; interested in biomolecular (photo)chemistry, photoinduced electron transfer and prebiotic chemistry; avid runner;
https://iam.pwr.edu.pl/people/rafal-szabla
I'm a theoretical physicist at Durham University
Associate Professor @OIST, our group is interested in understanding the origin of protein function. mom, climber, skier, co-founder of @rozforum.bsky.social
Computational Chemist @ Francis Crick Institute. #WomenInSTEM
📍 London, UK.
PhD at Leipzig University and TU Berlin working on machine learning for quantum chemistry
Reader in Energy Materials
Ion Transport and Interfaces Group www.jamesadawson.co.uk 🔋🌞🔥🖥
FACCTs is bringing the ORCA quantum chemistry software to industry - promoting the next Quantum leaps in the Pharma, Materials and Chemical Industries.
Visit us at www.faccts.de
ML-ready datasets and benchmarks for drug discovery.
https://polarishub.io
Science lead at Open Force Field. Core developer at MDAnalysis. Has never met an opossum.
Comp chem, chemical biology, software development and science communication at Open Force Field @openforcefield.org @omsf.io
She/they/he 🩷💛🩵 🏳️🌈
Scientific Editor of @chemicalscience.rsc.org, the flagship journal of the Royal Society of Chemistry. Diamond open access! | bioinorganic chemist | PhD Stanford University, post-doc University of York | wife, mum, gardener, Christian | she/her
Not-for-profit Focused Research Organisation (FRO) turning disordered proteins into viable drug targets | London, UK | https://bindresearch.org/
Head of Compute / Co-founder @bindresearch.org | Simulations, Drug Design, Disordered Proteins
Math Assoc. Prof. (On leave, Aix-Marseille, France)
Teaching Project (non-profit): https://highcolle.com/
https://palermolab.com
Giulia Palermo Lab at the University of California, Riverside. Biophysicist passionate about Science & Art! In love with Nucleic Acids and Computational Science!