Amos was not only a giant of bioinformatics and biocuration, but one of the nicest people I've met in academia. His support and advice were invaluable when we were establishing @bgee.org, and I will always remember how warmly he welcomed us to @sib.swiss when I arrived in Switzerland 20 years ago.
02.12.2025 09:30 — 👍 2 🔁 1 💬 0 📌 0
Intro to Bedder – The Quinlan Lab
We are thrilled to announce the first official release (v0.1.8) of #𝗯𝗲𝗱𝗱𝗲𝗿, the successor to one of our flagship tool, #𝗯𝗲𝗱𝘁𝗼𝗼𝗹𝘀! Based on ideas we conceived of long ago (!), this was achieved thanks to the dedication of Brent Pedersen.
1/n
02.12.2025 02:28 — 👍 298 🔁 152 💬 5 📌 11
Displacement-Optimized Tanglegrams for Trees and Networks https://www.biorxiv.org/content/10.1101/2025.11.26.690634v1
29.11.2025 17:46 — 👍 0 🔁 1 💬 0 📌 0

https://link.springer.com/article/10.1007/s00239-025-10277-1
Conceptual overview of hierarchical orthologous groups. An example of one HOG, or gene family. A Species tree with four taxa: plant (green), fish (blue), human (orange), and mouse (yellow), each with one or more genes. B The implied gene tree, dubbed “HOG tree,” and inferred nested HOG composition. Duplication nodes (red) can be deduced based on the species tree topology and clusters of homologous genes at each level. Ancestral genes from which the HOGs descended are shown in gray. C HOGs returned at different taxonomic levels. Consider a gene family that was present in the last eukaryotic common ancestor (LECA). At this level, a single HOG encompasses all genes descending from that ancestral gene. At the Vertebrata level, this gene underwent duplication, leading to two distinct copies, i.e., HOGs. At the Mammalia level, a second duplication further subdivides one of these HOGs, showing how deeper HOGs split into nested subHOGs at more recent levels. The HOG composition implies that a loss event occurred after the mammalian speciation

https://link.springer.com/article/10.1007/s00239-025-10272-6
Summary of the QfO8 meeting. a Hot topics and future directions in method development and applications within the QfO community, namely artificial intelligence, protein domains, protein structure, RNA and splicing isoforms. b Definition of orthology and paralogy, including various paralogous subtypes (e.g. in-paralogs and out-paralogs). c Duplications and functional divergence. d Applications of orthology

https://link.springer.com/article/10.1007/s00239-025-10271-7
Overview of the OrthoXML File Format (simplified). A schematic representation of an OrthoXML file, a standardized XML-based format for representing orthology data. OrthoXML follows a hierarchical structure where elements are enclosed within opening < tag > and closing </tag > tags. < orthoXML > is the root element enclosing other elements. The < species > element contains information about genes. An OrthoXML file can include a < taxonomy > element, which specifies the species tree used to generate the file. Additionally, the < groups > element encapsulates the orthology and paralogy relationships among genes
Our trilogy of orthology publications is online!
Review on Hierarchical Orthologous Groups doi.org/10.1007/s00239-025-10277-1
OrthoXML-Tools doi.org/10.1007/s00239-025-10271-7
A great community effort on Quest for Orthologs in the era of Data Deluge and AI doi.org/10.1007/s00239-025-10272-6
21.11.2025 16:26 — 👍 19 🔁 10 💬 1 📌 0
Great work by Nicola De Maio and Nick Goldman - not just scaleable to "pandemic scale" trees but - if I have got this right - arguably more valid than traditional column based bootstrap in the context of very tight evolution.
05.11.2025 22:33 — 👍 10 🔁 2 💬 0 📌 0

Yes - isometric scaling as a way to understand the benefits and costs of being small versus large. Haldane's Harpers article from 1926 is an amazing example of popular science writing.
31.10.2025 21:53 — 👍 22 🔁 6 💬 1 📌 0

Can an AI tool help us better understand the origins of cancer?
Researchers from EMBL's Korbel Group have developed a new AI method – MAGIC – which, through a game of molecular laser tag, is shedding light on how chromosomal abnormalities form in cells.
www.embl.org/news/science...
29.10.2025 16:11 — 👍 13 🔁 5 💬 0 📌 0

Unlocking the regulatory code of RNA: launching the Human RNome Project | Genome Biology | Full Text
The human RNome, the complete set of RNA molecules in human cells, arises through complex processing and includes diverse molecular species. While research traditionally focuses on four canonical nucleotide residues, the RNome, encompassing over 180 distinct modifications across organisms, with at least 50 in humans, is increasingly recognized. These modifications play critical roles in regulating RNA structure, stability, and function, yet the rules linking their precise locations to biological outcomes remain poorly defined. The Human RNome Project aims to map all RNA modifications, build essential resources, and harness new technologies to transform RNA biology, therapeutic development, agriculture, and even data storage.
Unlocking the regulatory code of #RNA: launching the Human #RNome Project genomebiology.biomedcentral.com/articles/10....
26.10.2025 18:11 — 👍 0 🔁 0 💬 0 📌 0
I am genuinely impressed by large language models - they can absorb disparate components of text into some consolidated view, they can produce extremely good language and - with the right model - translate pretty well between languages and they are an excellent text based UI for humans to use. But..
26.10.2025 07:48 — 👍 73 🔁 22 💬 2 📌 9

OpenAI and Anthropic v app developers: tech’s Cronos syndrome
Will the labs devour the apps that run on their models?
Think of AI labs as Cronos, a titan in Greek mythology, trying to devour his children. The question, as with Cronos, is: can the little ones survive and fight back?
25.10.2025 10:40 — 👍 2 🔁 3 💬 0 📌 0
Really exciting that the preprint on Barbell, a new demultiplexer, is finally out!
It's the first tool that builds on Sassy, the approximate-DNA-searching tool that @rickbitloo.bsky.social and myself developed earlier this year, specifically with this application in mind.
23.10.2025 21:28 — 👍 20 🔁 15 💬 2 📌 0
Full comic here: www.smbc-comics.com/comic/signal-4 #smbc
20.10.2025 17:16 — 👍 211 🔁 33 💬 9 📌 5

Nucleic acids and proteins
Big complex molecules are the unique stuff of life. This is how they work
Biological life depends on two families of large molecule: nucleic acids and proteins. The first of our collection of primers explains what they are and how they work
11.10.2025 12:20 — 👍 15 🔁 4 💬 1 📌 1
I am looking to get my hands on some #Illumina 5-base methylation data - does anyone have a bam file that I could use for some testing? Please RT for reach!
21.10.2025 09:36 — 👍 3 🔁 5 💬 2 📌 0

Illustration of Burrows-Wheeler Transform and many auxiliary structures from the input string how$now$brown$cow$#
New tool "bwt-svg" for making illustrations of the BWT and the many auxiliary arrays and other structures related to it. Pyodide-based no-installation-necessary interface here: benlangmead.github.io/bwt-svg/. (H/t to @robert.bio for pointing me to pyodide!) Full repo: github.com/benlangmead/....
14.10.2025 20:48 — 👍 40 🔁 21 💬 4 📌 1
Claus Wilke on Alphafold and the problem of protein folding in 2025
13.07.2025 20:41 — 👍 18 🔁 6 💬 0 📌 0

Go 1.25 interactive tour
Fake clock, new GC, flight recorder and more.
Go 1.25 interactive tour
Go 1.25 is scheduled for release in August, so it's a good time to explore what's new.
#golang
antonz.org/go-1-25/
28.06.2025 03:15 — 👍 6 🔁 2 💬 0 📌 0
Excited to launch our AlphaGenome API goo.gle/3ZPUeFX along with the preprint goo.gle/45AkUyc describing and evaluating our latest DNA sequence model powering the API. Looking forward to seeing how scientists use it! @googledeepmind
25.06.2025 14:29 — 👍 220 🔁 82 💬 5 📌 10

A general substitution matrix for structural phylogenetics.
Abstract. Sequence-based maximum likelihood (ML) phylogenetics is a widely used method for inferring evolutionary relationships, which has illuminated the
New paper from the lab from Sriram Garg in my group. We introduce a general substitution matrix for structural phylogenetics. I think this is a big deal, so read on below if you think deep history is important. academic.oup.com/mbe/advance-...
11.06.2025 14:01 — 👍 96 🔁 52 💬 3 📌 2
Powerful stuff from @juliosaezrod.bsky.social who found himself on the other end of the process - as a patient not a computational biology researcher - giving him insight into both research and patient perspectives. Huge credit to Julio for talking about his experiences here
20.06.2025 08:06 — 👍 31 🔁 10 💬 0 📌 0

Michael Ashburner FRS was an influential figure in the fields of Drosophila genomics and early sequencing database initiatives such as @ebi.embl.org.
Read about their contributions across genetics and bioinformatics in the new biographical memoir: buff.ly/f01zNat
@geneticscam.bsky.social
17.06.2025 10:44 — 👍 28 🔁 19 💬 3 📌 0
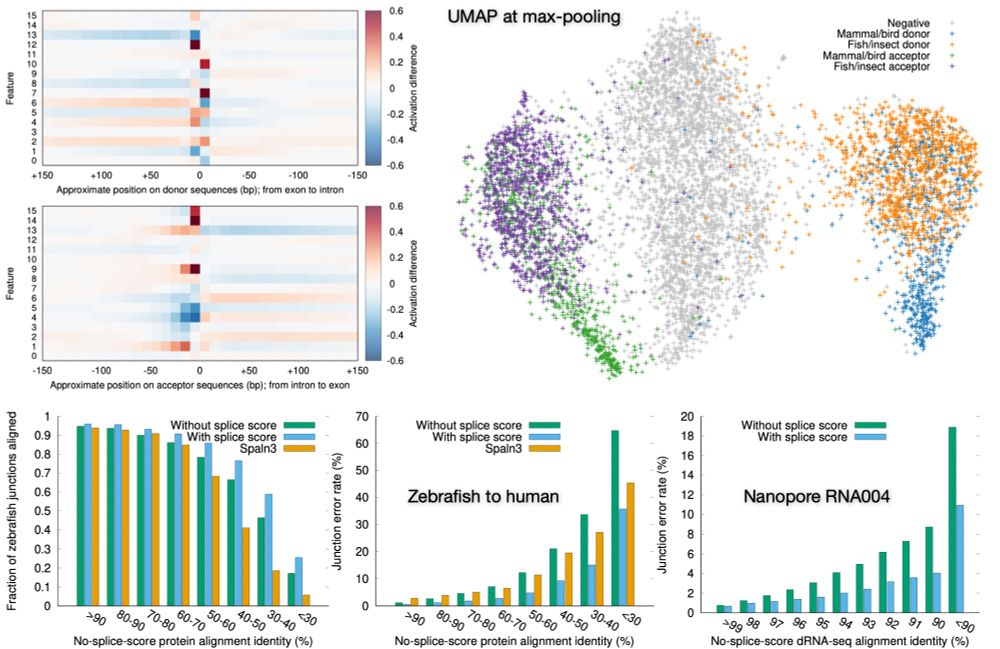
Preprint on "Improving spliced alignment by modeling splice sites with deep learning". It describes minisplice for modeling splice signals. Minimap2 and miniprot now optionally use the predicted scores to improve spliced alignment.
arxiv.org/abs/2506.12986
17.06.2025 01:48 — 👍 112 🔁 54 💬 0 📌 2
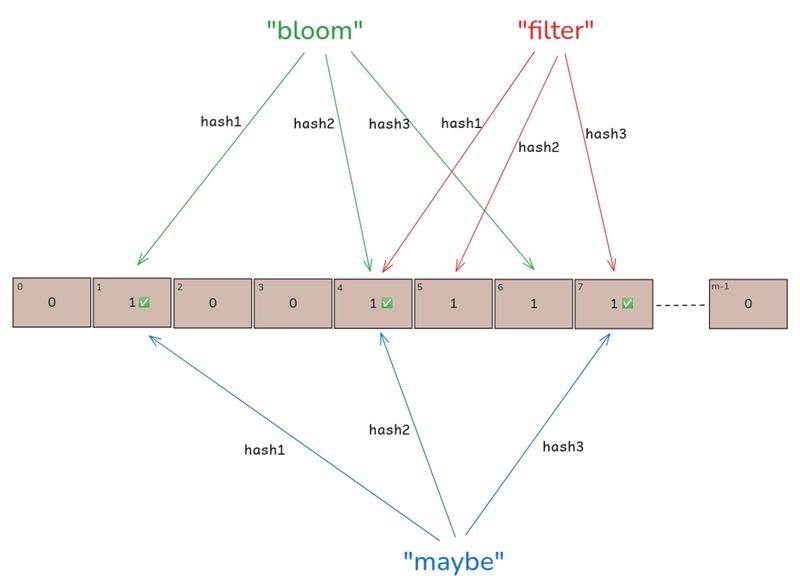
Probabilistic Data Structures in Go: Building and Benchmarking a Bloom Filter
#golang
dev.to/umangsinha1...
14.06.2025 04:29 — 👍 2 🔁 1 💬 1 📌 0
The App for Connecting Open Social Web
Mastodon, Bluesky, Nostr, Threads in ONE app, in ONE feed ✨
https://openvibe.social
Origins and consequences of genome mutation; software for genomic discovery.
Prof. and Chair of Human Genetics at U. of Utah.
https://www.genetics.utah.edu/
http://quinlanlab.org
Bloomberg Distinguished Professor of BME, CS, and Biostats at Johns Hopkins Univ., tennis player, @StevenSalzberg1 on Twitter, lab: salzberg-lab.org, Substack blog: stevensalzberg.substack.com
Author of Neuromancer and, most recently, Agency (still above from “Kill Switch”, The X-Files, Season 5, Episode 11, co-written with Tom Maddox)
Bioinformatician/Genomics Software Engineer @garvaninstitute.bsky.social
Views my own.
Mastodon @Psy_Fer_@genomic.social, https://genomic.social
Solve difficult biological problems using ML/AI.
Cancer Risk Assessment - “Catch Cancer Early”.
Perturbation Biology - computational cell biology - design new combo therapies.
Protein design - function and structure.
Developing data intensive computational methods • PI @ Seoul National University 🇰🇷 • #FirstGen • he/him • Hauptschüler
Chair, Computational BIology and Medicine Program, Princess Margaret Cancer Centre, University Health Network.
Associate Professor, Medical Biophysics, University of Toronto.
Disclosures: https://github.com/michaelmhoffman/disclosure/
Professor of Computer Science @ JHU. https://www.langmead-lab.org/ https://www.youtube.com/BenLangmead
Dad, husband, and PhD 🧑🧑🧒🧒👨🏻🎓 Scientist, technologist, and engineer 👨🔬📱👨💻 Bibliophile 📚 Philomath 🤓 Mental health advocate 💚
Passionate about #bioinformatics, #computing, #evolution, #genomics, #medicine, and #sre 🧬🩺💻
Homepage: https://www.gawbul.io
Genomics initiative lead at @GoogleDeepMind.
Models from our team: Enformer, AlphaMissense, and AlphaGenome.
Population and evolutionary genetics @UCDavis. Posts, grammar, & spelling are my views only. He/him. #OA popgen book https://github.com/cooplab/popgen-notes/releases
A bioinformatics postdoc at Victoria Popic's lab at Broad Institute.
Researcher in @BonsaiSeqBioinfo
Lille, France. Bioinformatics, data-structures for DNA/RNA.
https://x.com/golangch with over 30k followers
Go Development and AI Consulting: https://altafino.com
Clinical Microbiology #Bioinformatics @ Karolinska Hospital | PhD alum http://pharmb.io | Author of scipipe.org | Looking to get more into genomic algorithms & systems understanding of microbes+hosts, TEs etc
Married to Emebet. Dad. Sinner saved by grace.