What a cool way to discuss science. I have to watch this journal pub soon.
22.11.2025 23:01 — 👍 0 🔁 0 💬 0 📌 0
Do you or your lab use the Skyline software for #MassSpectrometry #proteomics? I'm looking for instructors to help with this year's Skyline Online, a virtual workshop/crash-course for all things Skyline! I'm especially looking for early career researchers for this opportunity. Please DM!
06.09.2025 17:11 — 👍 10 🔁 12 💬 0 📌 0
Did he get an intro like the one in 2018 US HUPO? "The one and only Hanno Steen"
30.04.2025 23:39 — 👍 1 🔁 0 💬 1 📌 0
With the #ASMS2025 app now available, here is a quick summary of what I've found so far from the abstracts:
Sciex - New ZenoTOF 8600, Echo-DMS
Thermo - New Exploris, New Astral (one of these is named Excedion), New OptiFlow source
Agilent - New single quad
Bruker - OmniTIMS, new triple quad, new LC
21.04.2025 20:06 — 👍 28 🔁 7 💬 4 📌 0
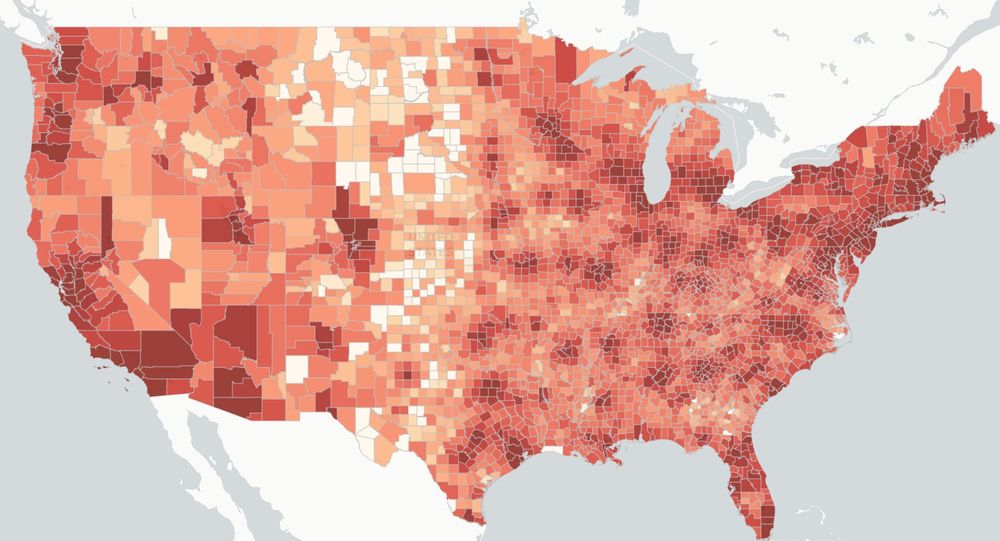
US map via scienceimpacts.org visualization of economic loss due to IDC cuts to 15% as part of Feb 7, 2025 executive order, with shading denoting intensity of cuts.
Working with an interdisciplinary team, we have developed a website to communicate how the White House's proposed cuts to health research would cause losses of $16B and 68,500 jobs.
Find out how your community may be impacted.
Explore more at SCIMaP: scienceimpacts.org
a 🧵
28.03.2025 02:15 — 👍 6499 🔁 3537 💬 200 📌 264

CDC plans to study autism, vaccines
Move comes despite strong evidence finding no link between them.
By LENA H. SUN and LAUREN WEBER • The Washington Post
I got your waste, fraud, and abuse right here
08.03.2025 14:26 — 👍 38029 🔁 7197 💬 1113 📌 536

One of the most powerful moments of the day came from Emily Whitehead who told the story of how she was the first pediatric patient to receive CAR T-cell therapy for her leukemia at age 5: “I stand up for science because science saved my life. And that’s a fact.” @standupforscience.bsky.social
08.03.2025 13:18 — 👍 764 🔁 182 💬 7 📌 2

Join the #StandUpForScience March because science is for everyone.
03.03.2025 22:00 — 👍 13 🔁 8 💬 0 📌 0

the word irony is written on a white background
ALT: the word irony is written on a white background
I have been trying to find the time to move away from the polical hellscape we find ourselves in to finish and share a bluetorial about science.
This helps me remember what this is all about.
Ironically, it is about the treatment of pain.
22.02.2025 10:19 — 👍 332 🔁 90 💬 10 📌 35

News in Proteomics Research blog post | Holy crap - US HUPO starts this week (Saturday! 2/22/25)! proteomicsnews.blogs...
---
#proteomics #prot-other
16.02.2025 16:40 — 👍 1 🔁 1 💬 0 📌 1

Real High Throughput Chemistry Through Mass Spec
Mass spec continues its relentless world takeover:
13.12.2024 19:36 — 👍 61 🔁 12 💬 1 📌 4
On the Hunt for the Histone Code
In BriefIn the early 2000s before high resolution mass spectrometers were readily available, the Hunt lab developed many approaches to analyze histone modifications on low resolution instruments.
In the early 2000s, Don Hunt's lab at UVa started developing mass spec based approaches to characterize histone PTMs. I and others were fortunate to be at the right place at the right time back then. Read an account about those early days here.
www.mcponline.org/article/S153...
12.12.2024 14:30 — 👍 64 🔁 17 💬 1 📌 1
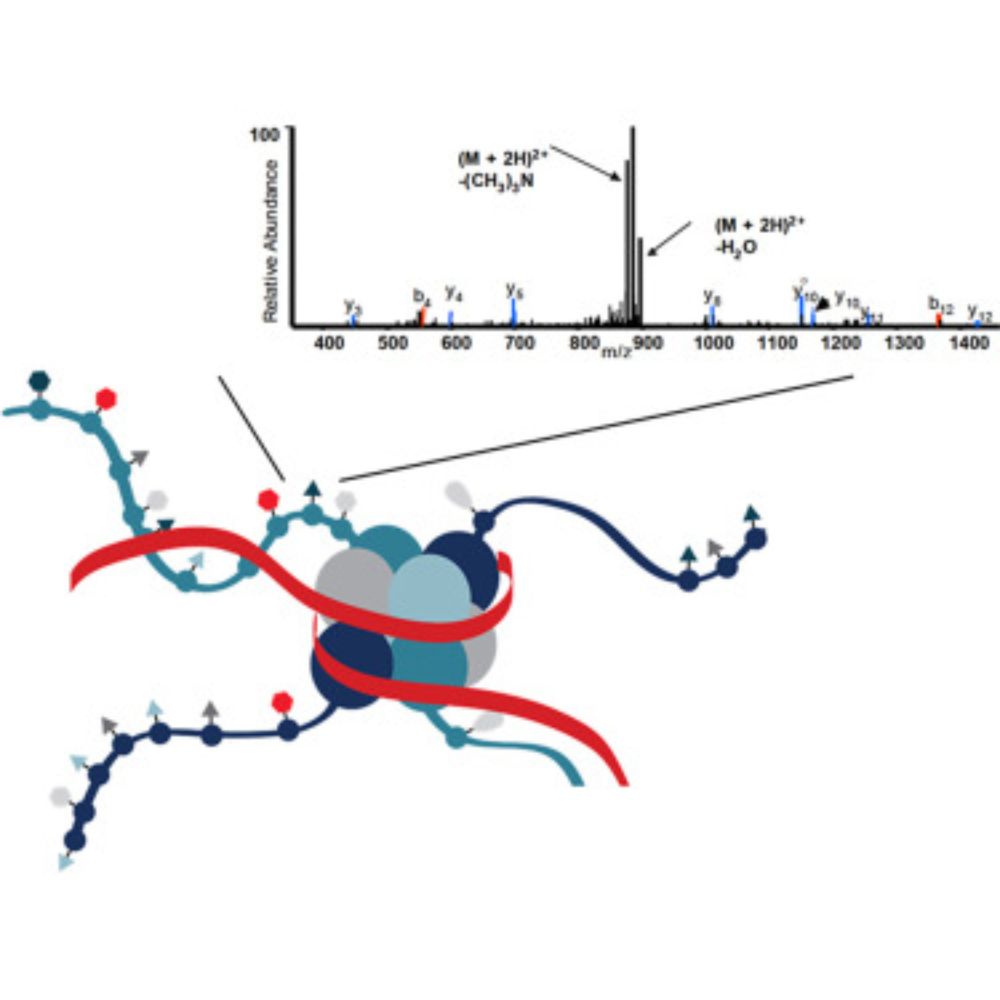
A Donald F. Hunt Story (John’s Version)
In BriefA personal narrative is provided on Donald Hunt’s development of tandem mass spectrometry methods for sequencing peptides.
Quite an exciting read.. everything we're doing today , from instrumentation to analytical methods (2DGE/ IMAC) to recent developments and interest in immunopeptidomics..all had roots in one place. An origins story told very well!
www.mcponline.org/article/S153...
10.12.2024 06:59 — 👍 1 🔁 1 💬 0 📌 0

Open-source and FAIR Research Software for Proteomics
Scientific discovery relies on innovative software as much as experimental methods, especially in proteomics, where computational tools are essential for mass spectrometer setup, data analysis, and in...
Recently, We saw a discussion on the role of open-source in proteomics. Here, experienced developers & researchers maintaining OS tools for years shared this comment to guide newcomers in the field about OS and its role in the field. 💻 #Proteomics #OpenSource chemrxiv.org/engage/chemr...
09.12.2024 13:03 — 👍 42 🔁 23 💬 1 📌 4

Tomorrow is the last day to apply! If you're seeking family and child care support for the annual conference, don’t forget to apply for the US HUPO 2025 Family and Child Care Support Award. Visit ow.ly/m7uE50U5IAu for more details. #USHUPO2025
05.12.2024 17:00 — 👍 1 🔁 2 💬 0 📌 0

Improved detection of differentially abundant proteins through FDR-control of peptide-identity-propagation
Quantitative analysis of proteomics data frequently employs peptide-identity-propagation (PIP) — also known as match-between-runs (MBR) — to increase the number of peptides quantified in a given LC-MS/MS experiment. PIP can routinely account for up to 40% of all quantitative results, with that proportion rising as high as 75% in single-cell proteomics. Therefore, a significant concern for any PIP method is the possibility of false discoveries: errors that result in peptides being quantified incorrectly. Although several tools for label-free quantification (LFQ) claim to control the false discovery rate (FDR) of PIP, these claims cannot be validated as there is currently no accepted method to assess the accuracy of the stated FDR. We present a method for FDR control of PIP, called “PIP-ECHO” (PIP Error Control via Hybrid cOmpetition) and devise a rigorous protocol for evaluating FDR control of any PIP method. Using three different datasets, we evaluate PIP-ECHO alongside the PIP procedures implemented by FlashLFQ, IonQuant, and MaxQuant. These analyses show that PIP-ECHO can accurately control the FDR of PIP at 1% across multiple datasets. Only PIP-ECHO was able to control the FDR in data with injected sample size equivalent to a single-cell dataset. The three other methods fail to control the FDR at 1%, yielding false discovery proportions ranging from 2–6%. We demonstrate the practical implications of this work by performing differential expression analyses on spike-in datasets, where different known amounts of yeast or E. coli peptides are added to a constant background of HeLa cell lysate peptides. In this setting, PIP-ECHO increases both the accuracy and sensitivity of differential expression analysis: our implementation of PIP-ECHO within FlashLFQ enables the detection of 53% more differentially abundant proteins than MaxQuant and 146% more than IonQuant in the spike-in dataset. ### Competing Interest Statement The authors have declared no competing interest.
Re-posting our new preprint on match between runs. This multi-lab effort (Keich, Noble, Payne & Smith) led by Alex Solivais should be of interest to anyone doing LFQ. We describe here how to control FDR in LFQ and provide the open source software to do it.
www.biorxiv.org/content/10.1...
02.12.2024 17:05 — 👍 31 🔁 15 💬 2 📌 2
You can use pyrrolysine.
24.11.2024 23:52 — 👍 1 🔁 0 💬 0 📌 0
My name includes 5 letters not coding for an amino acid. So, no MS2 spectra for me. 😔
24.11.2024 13:38 — 👍 1 🔁 0 💬 1 📌 0
bsky.app/profile/josu...
24.11.2024 13:24 — 👍 2 🔁 0 💬 0 📌 0
Last year, we had a thread on how our turkeys turned out. Maybe there's enough folks for a Mass Spec and BBQ starter pack. 😁
24.11.2024 12:49 — 👍 1 🔁 0 💬 1 📌 0
I'll be using my traeger. I often use hickory when smoking chicken and beef together. Oak for beef alone. And I think I used fruit wood for last year's turkey.
24.11.2024 12:34 — 👍 1 🔁 0 💬 1 📌 0
I've been catching up on the podcast during my morning commutes. This episode might just be the best commute.
24.11.2024 00:19 — 👍 0 🔁 0 💬 0 📌 0
I'm also smoking a turkey breast. What's you're go-to wood for smoking turkey?
24.11.2024 00:13 — 👍 0 🔁 0 💬 2 📌 0
Wow, this is great. Thanks.
22.12.2023 03:13 — 👍 2 🔁 0 💬 0 📌 0
This is a good question to ask Llyod Smith. I haven't seen him here, though.
22.12.2023 01:21 — 👍 1 🔁 0 💬 1 📌 0
Doing proteomics until I still can.
https://github.com/41ison
I research how spatial patterns of energy fuel the development of organs. BeziaLemma.com , listen to the Lemma Journal Club! https://www.youtube.com/@TheLemmaJournalClub
Transforming life sciences with cutting-edge #proteomics technology and solutions
Assistant Professor | Department of Chemistry, University of Minnesota | Biological Mass Spectrometry | Views are my own | http://gadkari.chem.umn.edu
Research and development at the intersection of cyberspace, global security, and human rights. Based at Munk School of Global Affairs & Public Policy, University of Toronto.
Econ professor at Michigan ● Senior fellow, Brookings ● Intro econ textbook author ● Think Like An Economist podcast ● An economist willing to admit that the glass really is half full ● Find me: https://linktr.ee/justinwolfers
Medicinal chemist / chemical biologist, author of “In the Pipeline” at http://science.org/blogs/pipeline. derekb.lowe@gmail.com and on Signal at Dblowe.18
All opinions are mine; I don’t speak for my employer in any way.
Assistant Director, Chemoproteomics at the Nucleus, Sarafan ChEM-H, Stanford University
Utrecht University, ETH Zürich and University of Vienna alumna.
#TeamMassSpec #StructuralProteomics #Proteomics #StanfordProteomics
🇦🇹➡️🇨🇭➡️🇳🇱➡️🇺🇸
MSAID transforms the way scientists analyze proteomics data
🌐 https://www.msaid.de
➡️ Request a Free CHIMERYS Demo: https://www.msaid.de/chimerys
Follow us: https://linktr.ee/msaid_de
#TeamProteomics #TeamMassSpec
SCIEX Mass Spectrometry Territory Manager - Eastern PA & DE
UD Alumni. Go Blue Hens!
Scientist | Discovery of unsaturated fatty acids, LC-MS lipidomics, precision photochemistry, organic & macromolecular chemistry
bioanalytical chemist | director of the metabolomics and proteomics core at unc-chapel hill | love PTM’omics and chemoproteomics | metabolomics cheerleader
Infectious disease physician, co-inventor of a rotavirus vaccine, author and grandfather. Author of "Tell Me When it's Over". Commentaries can be found on Pauloffit.substack.com.
NYU Law; MSNOW legal analyst & podcast cohost "Main Justice" (previously "Prosecuting Donald Trump"); NYT bestselling author.
Substack "Behind The Headlines" https://weissmann.substack.com
Offering legal analysis & opinion
brand new asst prof at University of Pennsylvania
studying metabolic flux- NCI R00 awardee- also i do bad jokes (she/her)
https://bartmanlabpenn.squarespace.com/
@ms.now senior legal reporter & recovering litigator; former off-air legal analyst @maddow.msnbc.com.
Don’t let the pearls fool ya.
Professor in Biomolecular Mass Spectrometry and Proteomics, Utrecht University, the Netherlands
The AI-powered developer platform to build, scale, and deliver secure software.
Mass spectrometrist helping deliver safer and more effective biopharmaceuticals
























