
Ben Langmead @benlangmead.bsky.social delivers the official opening for this year's Genome Informatics Conference #GI2025 at Cold Spring Harbor Laboratory.
List of talks and posters: meetings.cshl.edu/abstracts.as...

@mohsenzakeri.bsky.social
Postdoctoral Fellow at Johns Hopkins University, Computational Biology ❤️ www.mohsenzakeri.com
Ben Langmead @benlangmead.bsky.social delivers the official opening for this year's Genome Informatics Conference #GI2025 at Cold Spring Harbor Laboratory.
List of talks and posters: meetings.cshl.edu/abstracts.as...

Driveway at Cold Spring Harbor Lab, with conference attendees walking to a meal

Dolan Hall

Pic from a nighttime stroll
As our beloved Genome Informatics 2025 (#gi2025) approaches, I'm moved to share some photos from past years at CSHL. A couple more photos coming in a reply below...
03.11.2025 20:47 — 👍 19 🔁 6 💬 2 📌 1We’re all so proud of Steven Tan! He’s a super talented undergraduate at JHU. Thanks to the incredible support of @benlangmead.bsky.social, I had the opportunity to help guide this work, and I really loved it. Many thanks also to @sinamajidian.bsky.social for his valuable collaboration and input.
02.11.2025 19:27 — 👍 5 🔁 1 💬 0 📌 0Very excited about Movi 2! Excellent work by Mohsen here. FYI, I have a series of 5 videos on the move structure starting with this one: youtu.be/REniD2dKf6A?...
21.10.2025 21:39 — 👍 18 🔁 5 💬 0 📌 0PMLs are "exact" matches, but not maximal (unlike MEMs or matching statistics). That's why we call them "pseudo" matching lengths. PMLs are upper bounded by matching statistics.
23.10.2025 14:38 — 👍 1 🔁 0 💬 0 📌 0Great work and congrats to all authors! 🥳 Can't wait to read the preprint. Just for reference: a Fulgor index ⚡ on the same HPRC collection takes 8.26 GB (not in its most succinct representation). Fulgor, however, can be regarded as a "lossy" method here since it is based on kmers.
22.10.2025 07:41 — 👍 4 🔁 2 💬 0 📌 0Thank you!! Although that comes at the expense of more cache misses. 🙈
22.10.2025 05:37 — 👍 1 🔁 0 💬 0 📌 0
PML was introduced in SPUMONI. For matching statistics, there is a second loop which clarifies how much of the matches are overlapping, so the maximal exact matches could be retrieved. You can find out more about that in the MONI paper: pubmed.ncbi.nlm.nih.gov/35041495/
22.10.2025 05:34 — 👍 1 🔁 0 💬 1 📌 0When the match is not extendable, the thresholds are still useful, because they point to the direction which has a longer common prefix with the current match. After each repositioning we always reset the PML to zero because we are not sure if it is extending a match or not. That’s PML.
22.10.2025 05:30 — 👍 1 🔁 0 💬 1 📌 0We would like to choose the one that has a longer common prefix with the current row because that is more likely to extend the match. The thresholds exactly encode this information to guide the search to the right direction, possibly to get back into the backward search range.
22.10.2025 05:26 — 👍 1 🔁 0 💬 1 📌 0PML proceeds by LF on one row, for the first two steps (CC) PML row remains in the backward search range, so the match len is extended. But then for G, we don’t see a matching character in the purple row. So, PML repositions either to the bottom of the preceding G run or the head of the next G run.
22.10.2025 05:25 — 👍 0 🔁 0 💬 1 📌 0
This figure from our Movi Color preprint could address your questions. (Don’t worry about the colors) Here we compare the PML query to backward search. The purple box shows the row tracked by PML and the green BWT offsets track the backward search intervals on the same query (AGCC).
22.10.2025 05:20 — 👍 1 🔁 0 💬 1 📌 0Exactly! In Movi we like to access the thresholds directly from the move rows, and we need it for each character different than the run's character (in the case of a mismatch during the PML query).
22.10.2025 05:16 — 👍 2 🔁 0 💬 0 📌 0
A formal dentition of the thresholds is in this paper (Refining the r-index): www.sciencedirect.com/science/arti...
22.10.2025 05:11 — 👍 1 🔁 0 💬 0 📌 0
6/6 Movi 2 supports multi-threading, further improving speed beyond the concurrent read processing per thread already available in Movi 1. You can read more about Movi 2 at: www.biorxiv.org/content/10.1...
21.10.2025 20:07 — 👍 1 🔁 0 💬 0 📌 0
5/6 On the 466 haplotypes from the 2nd release of HPRC, the fastest Movi 2 index is under 50 GB. It can be reduced to 24 GB while remaining over 3x faster than SPUMONI. Movi 2 is smaller and faster than ropebwt3, although it computes PMLs, which are easier to get than the SMEMs found by ropebwt3.
21.10.2025 20:07 — 👍 1 🔁 0 💬 2 📌 1
4/6 Movi 2 offers three main modes: regular, blocked, and sampled. Each mode uses a different row size, resulting in a different number of added rows due to its specific splitting strategy.
21.10.2025 20:07 — 👍 1 🔁 0 💬 1 📌 0
3/6 Movi 2 includes a mode that samples the largest field in each row to achieve a space–speed tradeoff. In this mode, it can be smaller than r-index–based methods while remaining 3–8× faster.
21.10.2025 20:07 — 👍 4 🔁 0 💬 2 📌 0
2/6 Movi 2 uses length-based and threshold-based splitting to reduce the size of rows in the move structure. The threshold-splitting strategy compresses each threshold value to a single bit.
21.10.2025 20:07 — 👍 3 🔁 1 💬 2 📌 0
1/6 Movi 2 is here: faster and more space-efficient for pangenome queries. Its fastest mode uses half the memory of Movi 1 while running ~30% faster. github.com/mohsenzakeri...
21.10.2025 20:00 — 👍 44 🔁 24 💬 1 📌 2An awesome opportunity, strongly recommended to anyone looking for a postdoc in algorithms and genomics!
09.10.2025 15:18 — 👍 5 🔁 1 💬 0 📌 1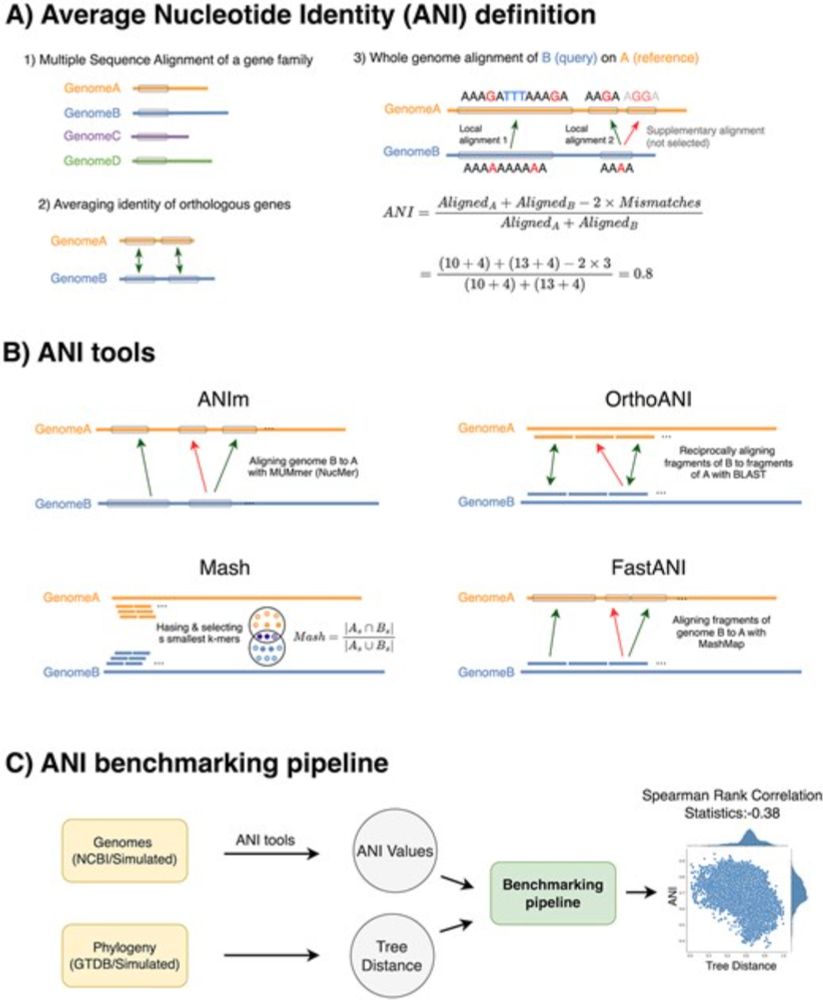
EvANI benchmarking workflow for evolutionary distance estimation academic.oup.com/bib/article/...
An great teamwork by @mohsenzakeri.bsky.social, @stephenhwang.bsky.social and me, with the excellent mentorship of @benlangmead.bsky.social

Anchor-based merging requires a common sequence (red) present in each partition. Multi-MUMs are merged by identifying overlaps between partition-specific matches in the anchor coordinate space, and a uniqueness threshold determines if a MUM is still unique in each partition after truncation. (B) String-based merging enables computation of multi-MUMs between partitions without a common sequence. An example tree (left) is shown, highlighting the use case where partial multi-MUMs specific to internal nodes (starred) can be computed by merging subclade- based partitions up a tree. (right) MUM overlaps are computed by running Mumemto on the MUM sequences, and the uniqueness threshold array ensures overlaps remain unique across the merged dataset. (C) An example Burrows-Wheeler Transform (BWT), matrix (BWM), and Longest Common Prefix (LCP) array, with sequence IDs for each suffix shown (ID). A non-maximal unique match (UM) is shown, and the uniqueness threshold for this match is found using the flanking LCP values. (D) A partial multi-MUM (in blue) is found in all-but-one sequence (excluded in red). Using two anchor sequences (red and orange), all-but-one partial MUMs can be computed using an augmented anchor-based merging method.


(A) Phylogeny of geographically diverse A. thaliana accessions (Lian et al. 2024), with broad geographical regions colored. Internal nodes are labeled with the coverage of partial multi-MUMs across the leaves of each node. Internal node partial MUMs are computed by merging subtree-based partitions progressively up the phylogeny. (B) Global multi-MUM synteny across the full dataset shown in blue (with inversions in green). Global MUMs are computed by merging all partitions together (representing the root node). Additionally, three geographically distinct subgroups are highlighted and partition-specific multi-MUMs (in purple, with inversions in pink) reveal local structural variation in centromeric regions.
Great talk by Vikram @vikramshivakumar.bsky.social on studying pangenomes and synteny visualization in #WABI25
Github: github.com/vikshiv/mume...
First paper: genomebiology.biomedcentral.com/articles/10....
Second: www.biorxiv.org/content/10.1... #WABI2025
The 25th iteration of the excellent Conference for Algorithms in Bioinformatics (WABI) starts tomorrow at UMD @umdscience.bsky.social at the Brendan Iribe Center. You can find details at the website wabiconf.github.io/2025/. We'll use the tag #WABI25 for the meeting!
20.08.2025 02:43 — 👍 17 🔁 9 💬 0 📌 0🧬🖥️ In addition to an update to oarfish, a new version (0.14.0) of piscem (zenodo.org/records/1509...) has just been released. This version pulls in some of the latest improvements to sshash by @jermp.bsky.social! 1/2
30.06.2025 14:14 — 👍 5 🔁 3 💬 1 📌 0The second keynote address at WABI '25 will be by Christina Boucher. She will talk about "Recursive Parsing and Grammar Compression in the Era of Pangenomics". PFP (& RPFP) has enabled tremendous advances in representation & indexing; this will be an exciting talk!
wabiconf.github.io/2025/talks/t...
No war with Iran
20.06.2025 00:22 — 👍 934 🔁 116 💬 50 📌 3Excited to introduce LiftOn – an open-source tool for accurate, scalable liftover of genome annotations (GFF) across assemblies. 🚀
👉 Code & community: github.com/Kuanhao-Chao...
It’s been incredibly rewarding building this for the genomics community. Can’t wait for your feedback and contributions!
Huge congrats! 🎉
07.06.2025 22:37 — 👍 1 🔁 0 💬 1 📌 0Excellent work, Steven & Mohsen! See thread below
29.05.2025 14:56 — 👍 8 🔁 2 💬 0 📌 0

















