Congratulations!!!!!!!!!!
07.06.2025 03:19 — 👍 1 🔁 0 💬 1 📌 0
Introns have to come from somewhere, right? @celineh2ooo.bsky.social and I looked at multiple genome alignments with 1000s of genomes and found 342 cases where humans (and our relatives) had gained a new intron. Still not sure where these come from, but it's a fascinating question
04.06.2025 20:13 — 👍 42 🔁 12 💬 2 📌 1
Neng Huang developed longcallR for joint SNP calling and phasing from long RNA-seq reads, AND for identifying allele-specific splicing/junctions (ASJ). Although ASJs of statistical significance are rare, a large fraction involve unannotated junctions. In Rust!
30.05.2025 14:54 — 👍 16 🔁 7 💬 0 📌 0
Industry friends, now is the time for MUCH more speaking out on behalf of academic colleagues under duress. Here are core open source methods that many of your products doubtlessly depend on either directly or indirectly (see en.wikipedia.org/wiki/HMMER) being abruptly defunded. Make noise.
29.05.2025 14:39 — 👍 74 🔁 49 💬 1 📌 0
myloasm - metagenomic assembly with (noisy) long reads
Announcing myloasm, a new long-read (ONT R10/PacBio) metagenome assembler that I've been working on during my postdoc in the Heng Li lab (@lh3lh3.bsky.social).
myloasm-docs.github.io
28.05.2025 17:53 — 👍 132 🔁 78 💬 5 📌 3

Partitioned Multi-MUM finding for scalable pangenomics
Pangenome collections are growing to hundreds of high-quality genomes. This necessitates scalable methods for constructing pangenome alignments that can incorporate newly-sequenced assemblies. We prev...
Excited to share a new update to Mumemto, scaling MUM and conserved element finding to any size pangenome! Preprint out now w/ @benlangmead.bsky.social.
Mumemto scales to the new HPRC v2 release and beyond, and can merge in future assemblies without any recomputation! 1/n
27.05.2025 19:35 — 👍 27 🔁 15 💬 1 📌 2
Centrifuger has updated the pre-built index list to include this exciting GTDB new release r226 for taxonomic classification of sequencing data: github.com/mourisl/cent.... There is also a gtdb+refseq human/virus/fungi/contaminants index, hopefully will be useful for human microbiome studies.
27.05.2025 15:58 — 👍 3 🔁 0 💬 0 📌 0
Great 🧵 by Pierre on the Kaminari paper! In short, Kaminari is a simple and elegant, but highly effective index for approximate colored k-mer queries. The simplicity leads to very fast query, but with accuracy consistent with (or exceeding) best-in-class solutions; a very fun collaboration indeed!
27.05.2025 15:41 — 👍 10 🔁 2 💬 0 📌 0
Congratulations!!!!
09.05.2025 21:14 — 👍 1 🔁 0 💬 1 📌 0
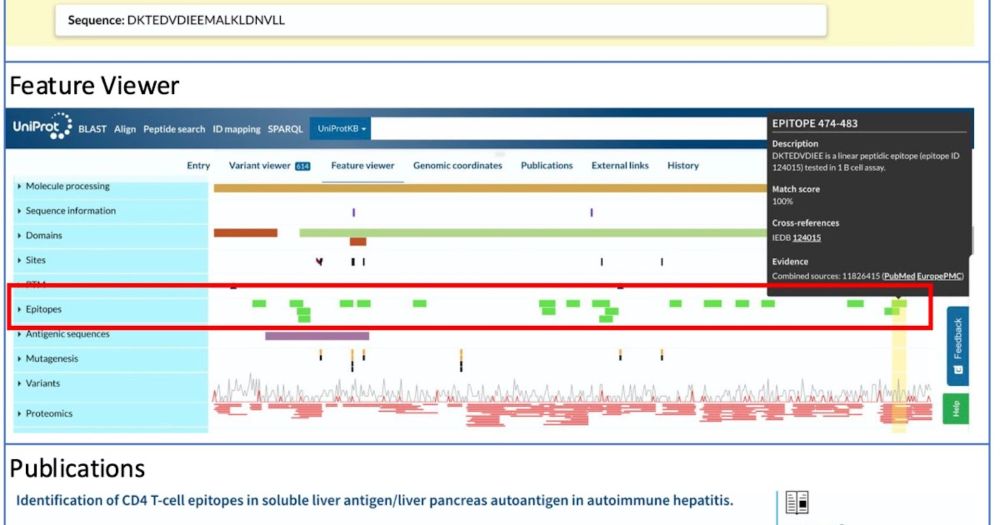
Inside UniProt
Rich Epitope Information Comes to UniProt Mammalian immune responses are mediated by interactions between antigens and immune system compo...
Check out our latest collaboration with UniProt, who has integrated over 700,000 experimentally validated epitopes to enhance its protein entries with detailed immune response information. This data is accessible via the UniProt Feature Viewer and API! 💻🔬🧪 #collaboration #immunology #proteins
09.05.2025 00:35 — 👍 2 🔁 1 💬 0 📌 0
WABI 2025
WABI Conference on Algorithms in Bioinformatics
The deadline for WABI 2025 has been extended (but is still rapidly approaching) wabiconf.github.io/2025/
* abstract deadline: May 12 (AoE)
* paper deadline: May 15 (AoE)
Consider submitting your exciting algorithmic bioinformatics work to the WABI conference!
07.05.2025 19:14 — 👍 10 🔁 11 💬 0 📌 2
Thank you!
04.05.2025 14:46 — 👍 0 🔁 0 💬 0 📌 0
Forgot to dustmasker the genomes before creating a Centrifuger index and indeed saw some misclassifications. Took a while to figure out and lessons learned... Need to implement a built-in masking step like Kraken2 in case forget doing it in the future..
04.05.2025 06:25 — 👍 0 🔁 0 💬 1 📌 0

GitHub - ArcInstitute/xsra: An efficient CLI to extract sequences from the SRA
An efficient CLI to extract sequences from the SRA - ArcInstitute/xsra
Extracting @NCBI SRA files with fasterq-dump can require 17x the size of the accession while decompressing. Our new tool xsra extracts sequences at 5x throughput with significantly less disk usage, built-in compression, and optional BINSEQ outputs
github.com/arcInstitute...
29.04.2025 21:03 — 👍 40 🔁 15 💬 2 📌 1
AllTheBacteria
Small update from AllTheBacteria (allthebacteria.org). Assemblies can be bulk downloaded from OSF as before, or you can now get individual assemblies from AWS. We now also have a LexicMap index on AWS, so you can align your favourite gene against 2.4million bacteria (next post for price estimates)
29.04.2025 15:36 — 👍 47 🔁 23 💬 1 📌 2

The Department of Human Genetics at the University of Utah is sponsoring the Rising Stars in Genetics and Genomics symposium!
- We are seeking nominations bu June 1.
- September 18-19, 2025
- Please share with the star postdocs that you know.
docs.google.com/forms/d/e/1F...
28.04.2025 17:20 — 👍 54 🔁 43 💬 1 📌 1

The sequence analysis session of #RECOMB2025 is off to a great start with @jimshaw.bsky.social presenting devider, a new algorithm for haplotyping small sequences from long-read sequencing.
www.biorxiv.org/content/10.1...
27.04.2025 01:27 — 👍 26 🔁 6 💬 1 📌 0

If you want to check if a human gene has copy-number changes or lands in a complex region, try pangene.bioinweb.org. Recently updated with more and better assemblies.
26.04.2025 01:06 — 👍 44 🔁 14 💬 2 📌 0
Time to build a new index!!
24.04.2025 01:54 — 👍 7 🔁 1 💬 0 📌 0
Short RNA-seq read alignment with minimap2
Minimap2-2.29 released with the support of short RNA-seq read alignment. More explanation and results here: lh3.github.io/2025/04/18/s...
18.04.2025 21:53 — 👍 29 🔁 7 💬 0 📌 0
Short RNA-seq read alignment with minimap2
minimap2 adds support for short read spliced RNA-seq alignment! lh3.github.io/2025/04/18/s...
18.04.2025 21:58 — 👍 34 🔁 8 💬 1 📌 1
Happy Birthdays, Ben and Rob! Very 2-power day!
02.04.2025 18:51 — 👍 2 🔁 0 💬 0 📌 0
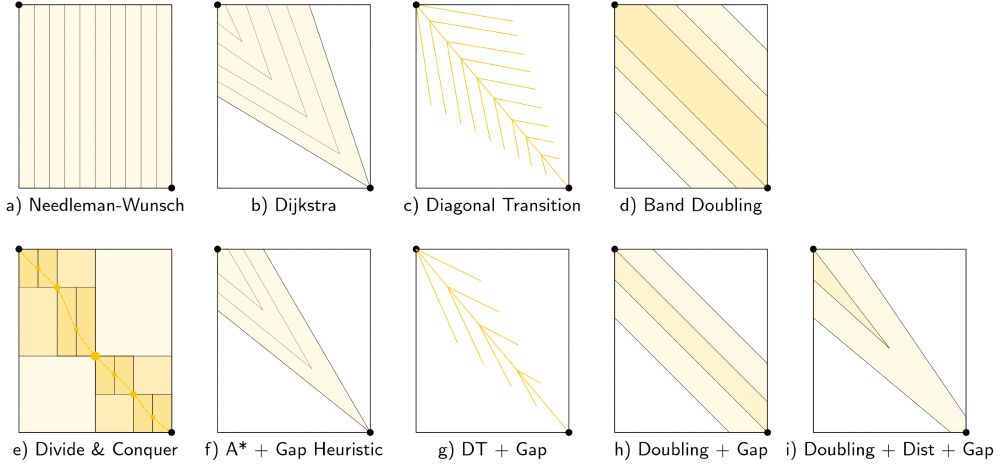
Schematic figures showing global pairwise alignment algorithms
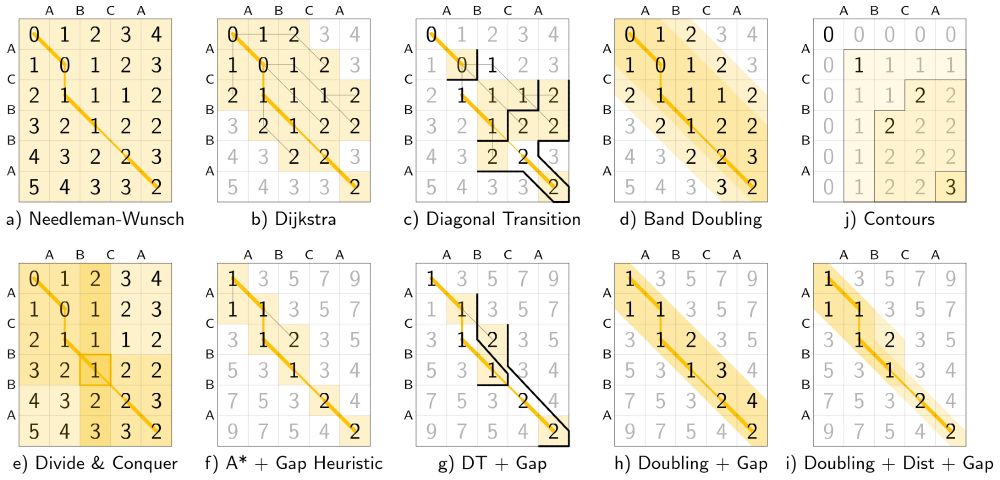
A worked example for each algorithm
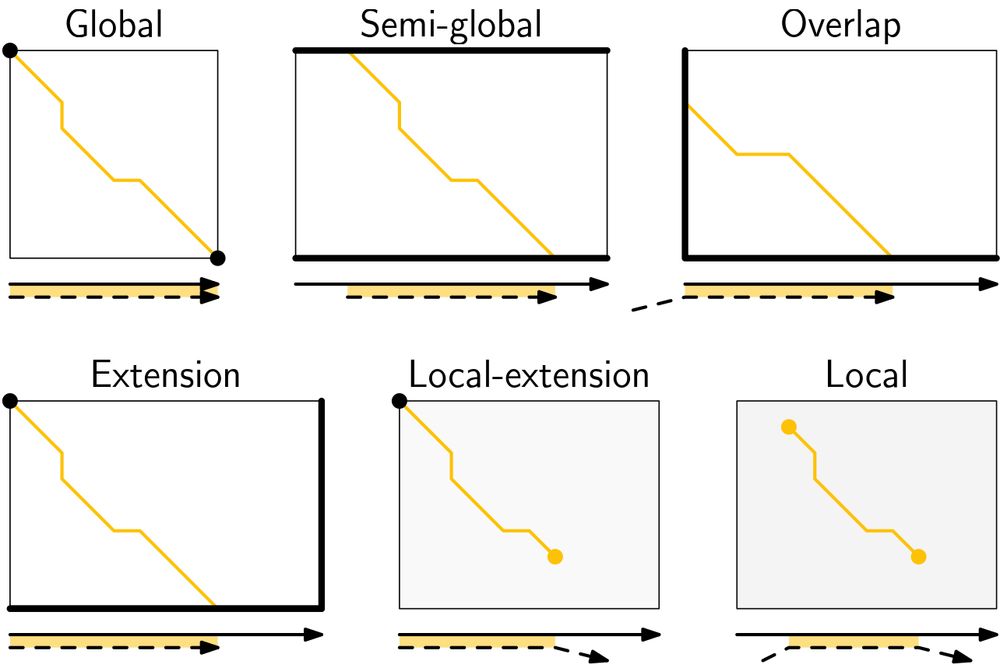
Schemetic figures showing various modes, such as semi-global, local, and extension alignment
New set of thesis figures on pairwise alignment just dropped!
- schematic and worked example for many algorithms
- alignment modes
27.03.2025 16:30 — 👍 33 🔁 9 💬 2 📌 0
GitHub - fulcrumgenomics/fqgrep: Grep for FASTQ files
Grep for FASTQ files. Contribute to fulcrumgenomics/fqgrep development by creating an account on GitHub.
fqgrep release 1.1.0 now speeds up searching FASTQ files!
Thank-you to both Markus Schlegel from @activegroupgmbh.bsky.social for updating seq_io and Nicholas D. Crosbie of grepq for some competition and inspiration.
See more: github.com/fulcrumgenom...
14.03.2025 17:45 — 👍 12 🔁 5 💬 1 📌 0
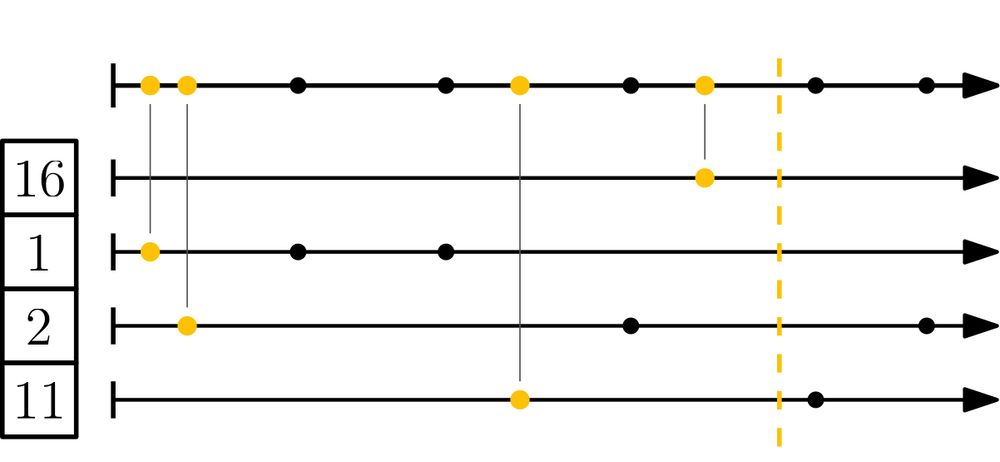
Schematic of the mod-bucket algorithm: all k-mer hashes are partitioned into s buckets via their remainder mod s. Then, in each bucket the smallest hash is selected.
Just published simd-sketch, a crate for fast bucket sketches.
It's 7x to 30x faster than BinDash, by using the simd-minimizers crate for fast hashing, and a nearly branch-free implementation.
Here's a blogpost with a survey of minhash history & methods, and evals:
curiouscoding.nl/posts/simd-s...
14.03.2025 00:35 — 👍 12 🔁 9 💬 1 📌 0
Scientist at the New York Genome Center & NYU.
http://sanjanalab.org
Foundational tools for omics data (mostly in python).
PI of @EvomicsLab.bsky.social (www.evomicslab.org)
at Sun Yat-sen University Cancer Center (SYSUCC)
Alumni of @cnrs.fr @ircan.bsky.social @RiceUniversity @NanjingUnivers1
AI Research @Hugging Face 🤗
Contributing to the Chinese ML community.
Associate professor of Bioinformatics at Chongqing Medical University, China. Lab: https://mbio.info, Personal: https://shenwei.me, https://x.com/shenwei356
Postdoc researcher in bioinformatics at Pasteur institute. Scalable methods and software for metagenomics. https://github.com/GaetanBenoitDev
@cshlnews.bsky.social postdoc with @hannahvmeyer.bsky.social and Saket Navlakha • How your T cells know it's you • Develops immuno tools named after Gotham characters: github.com/meyer-lab-cshl/BATMAN 🦇
Inria Senior researcher.
Head of the https://team.inria.fr/genscale/ at Inria and Irisa.
Algorithmics for sequencing data analyses, genomics and metagenomics.
Bioinformatician @ Basecamp Research
PHD student in Computational Mathematics @ Stockholm University, developing algorithms for long read sequencing data, github: https://github.com/aljpetri/
Professor, USC Marine and Environmental Biology, microbe hunter. he/him. Not a bot. thethrashlab.com
PIRL Co-PI (https://pirl.unc.edu/). Trying to figure out adaptive immunity to cancer, at least enough to develop better immunotherapies.
MD-PhD candidate @utmbhealth interested in viruses and data science | Former ballet dancer using publicly available dance data for advocacy
https://jason-yeung.netlify.app/
https://datapointesguide.com/
Celebrating the Arts and Sciences community and scholarship that pushes the boundaries of discovery and creativity. 🌲https://faculty.dartmouth.edu/artsandsciences/
Bioinformatics Scientist at the Arc Institute.
Working at the intersection of functional genomics, systems biology, and machine learning. I also build rusty bioinformatics tools
https://github.com/noamteyssier
My lab at Stanford studies human population genetics and complex traits.























