And a huge thank you to these incredible people!
@alwaysrong.bsky.social @sagergosai.bsky.social @rodrigoicastro.bsky.social @mackenzie-noon.bsky.social @pardissabeti.bsky.social @stevereilly.bsky.social @tewhey.bsky.social @yaleschoolofmed.bsky.social @jacksonlab.bsky.social @umaine.bsky.social
23.04.2025 17:28 — 👍 1 🔁 0 💬 0 📌 0
We have produced a comprehensive catalog of non-coding variant effects and make these predictions available to the community. We encourage researchers interested in gene regulation across fields to explore our precomputed predictions or generate their own to guide their future experiments!
23.04.2025 17:28 — 👍 1 🔁 0 💬 1 📌 0
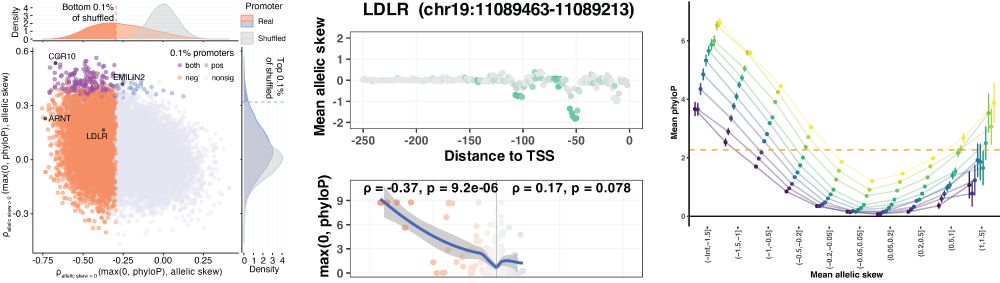
Lastly we investigate all human promoters by saturation mutagenesis, identifying canonical promoter TFs and linking non-coding variant effect size to coding constraint (LoEUF), bridging the gap between coding and non-coding function.
23.04.2025 17:28 — 👍 0 🔁 0 💬 1 📌 0
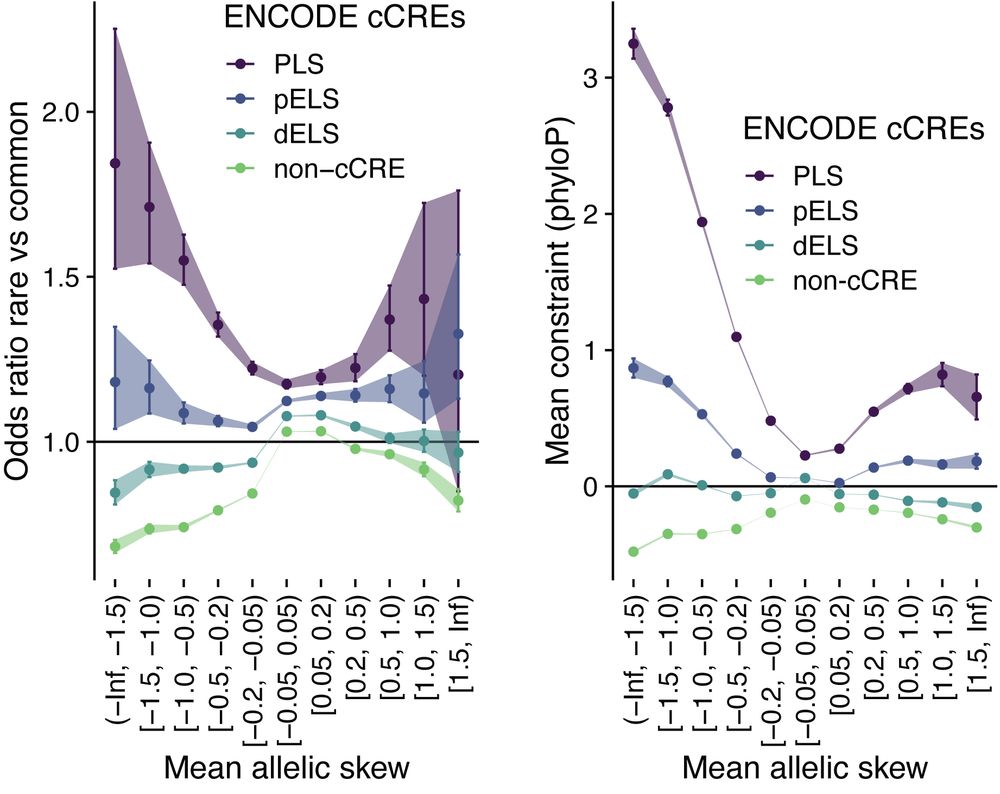
MPAC can scale to predict 514M gnomAD variant effects and we quantify the relationship between allele frequency or evolutionary conservation with predicted skew at an unprecedented level. Notably, we find that variants causing high skew are under greater constraint than those with small effects.
23.04.2025 17:28 — 👍 1 🔁 0 💬 1 📌 0
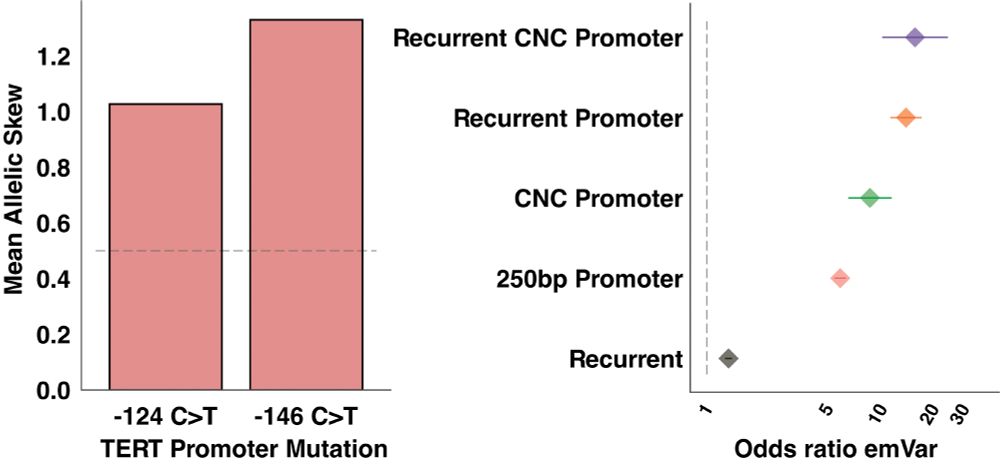
In COSMIC we identify known non-coding driver mutations (TERT) and by combining variant recurrence, regulatory element annotations, and cancer-associated promoters we nominate 1,892 emVars as putative non-coding drivers.
23.04.2025 17:28 — 👍 0 🔁 0 💬 1 📌 0
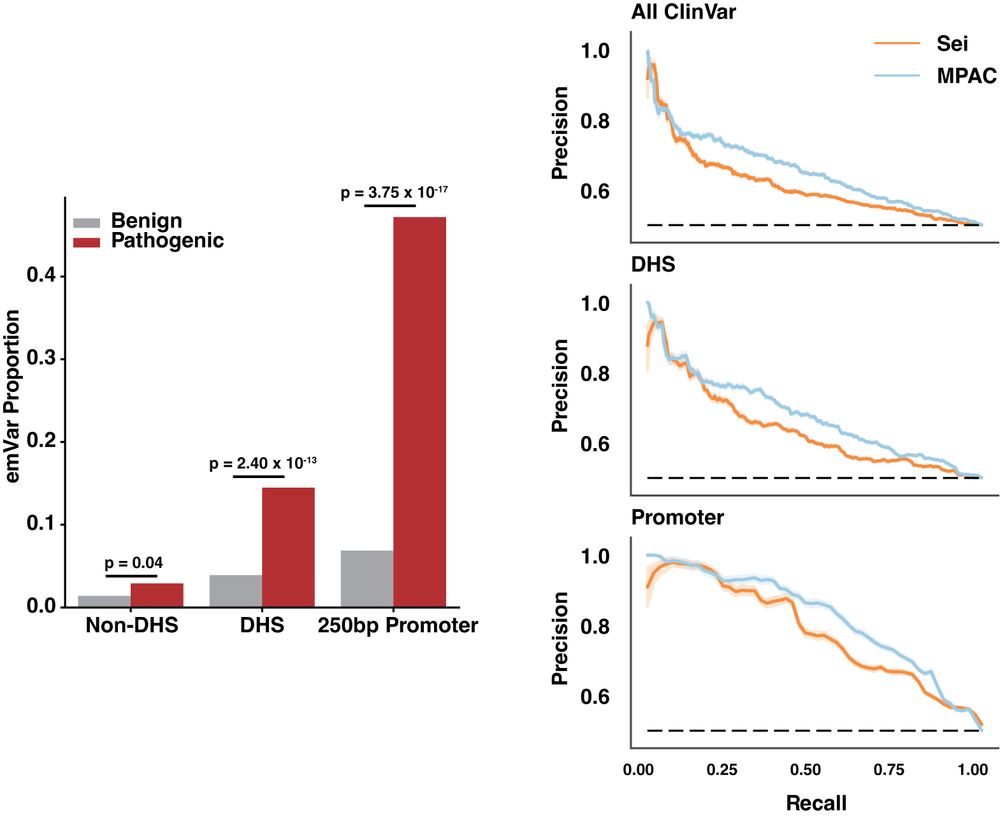
Many clinically identified non-coding variants lack clear effects, using MPAC we can predict the impact of all ClinVar non-coding variants and observe enrichments in pathogenic alleles for highly disruptive variants (emVars).
23.04.2025 17:28 — 👍 0 🔁 0 💬 1 📌 0
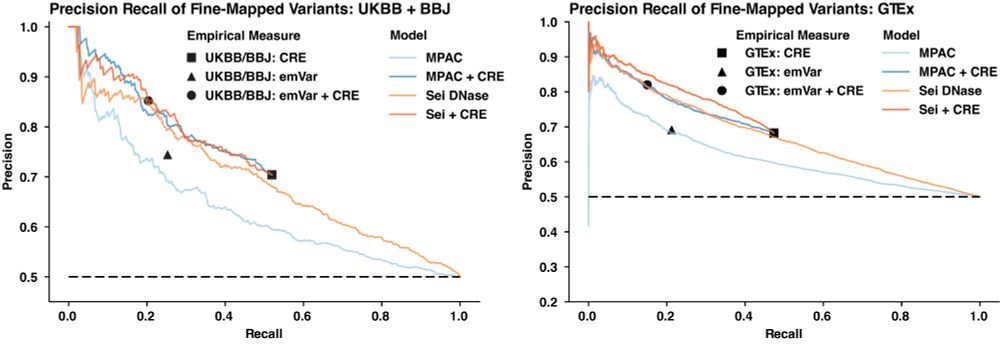
MPAC predictions distinguish causal variants from the UK Biobank, Biobank Japan and eQTLs from GTEx with experimental accuracy but without experimental overhead!
23.04.2025 17:28 — 👍 0 🔁 0 💬 1 📌 0
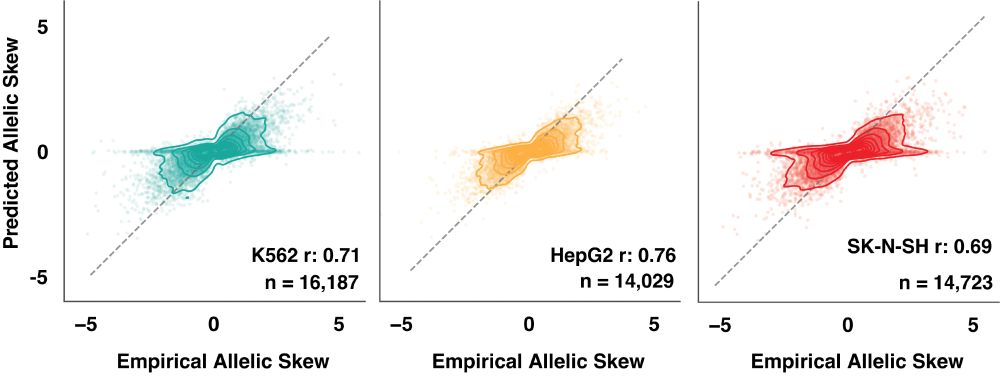
Trained on MPRA data from a large-scale study of human trait and eQTL variants (www.biorxiv.org/content/10.1...) and extending the Malinois model architecture (www.nature.com/articles/s41...) MPAC predicts variant effects with high accuracy.
23.04.2025 17:28 — 👍 0 🔁 0 💬 1 📌 0
Massively Parallel Reporter Assays (MPRAs) quantify the activity of 10-100s of thousands of sequences, however, it is not feasible to test all known variation. Modeling MPRA can increase scale and lead to better understanding of complex traits, somatic and germline diseases, and population genetics.
23.04.2025 17:28 — 👍 0 🔁 0 💬 1 📌 0
| bioRxiv
bioRxiv - the preprint server for biology, operated by Cold Spring Harbor Laboratory, a research and educational institution
Excited to share our MPAC preprint, a scalable ensemble of ML models for genome-wide non-coding variant effect prediction and our findings from 575M predictions across databases including @ukbiobank.bsky.social, GTEx, ClinVar, COSMIC, and @gnomad-project.bsky.social
www.biorxiv.org/content/10.1...
23.04.2025 17:28 — 👍 17 🔁 9 💬 1 📌 3
Black Sky Thinking since 2008.
Help support independent cultural journalism & get perks by becoming a tQ subscriber here: https://steadyhq.com/thequietus
Assistant Professor, UBC school of Biomedical Engineering. Trying to enable personalized medicine by solving gene regulatory code.
Professor of EECS and Statistics at UC Berkeley. Mathematical and computational biologist.
precision psychiatry & psychiatric genetics
postdoc @ massgen | harvard med | broad institute
Curiosity. Human Model Systems. Single-Cell Genomics. EvoDevo…and more!
Assistant Professor @ Stanford Genetics & BASE Initiative. Mapping the regulatory code of the human genome to understand heart development and disease. www.engreitzlab.org
Asst Prof at University of California, Irvine.
Genetics, Genomics, Gene Regulation, Development. Views are my own.
https://www.kvonlab.org/
Population geneticist, University of Chicago
Genomics, technology and human genetics @University of Washington. Working to create an atlas of variant effects and resolve VUS.
Stanford PhD candidate in the Engreitz Lab. Passionate about engineering gene regulation and high-throughput tech dev.
Sequence plasmids, amplicons, colonies, AAV, yeast, and bacterial genomes! Our advanced sequencing delivers comprehensive visibility, higher accuracy, and the ability to detect structural variations and rare mutations that other sequencing methods miss.
Laboratory of Professor James Noonan | Department of Genetics at Yale University. Science is a team sport. Reposts are not necessarily endorsements. We speak only for ourselves. Website: noonanlab.org
Senior Investigator @ Altius Institute for Biomedical Sciences. Research: High-resolution mapping of chromatin structure & function. Fun: Mountain shenanigans and skiing turns all year. Seattle, USA/Patagonia Chilena (🇺🇸🇨🇱). http://vierstra.org
Cell aims to publish the most exciting and provocative research in biology. Posts by Scientific Editors on the Cell editorial team. See the latest papers at https://www.cell.com/cell/
Group leader @crg.eu | blood, single cell, synthetic genomics
Professor and Diana Davis Spencer Foundation Chair, The Jackson Laboratory
Studying genomics, machine learning, and fruit. My code is like our genomes -- most of it is junk.
Assistant Professor UMass Chan, Board of Directors NumFOCUS
Previously IMP Vienna, Stanford Genetics, UW CSE.
Curating and cataloging the world's data for the fruit fly Drosophila melanogaster into our database at http://flybase.org. Aiding discovery since 1992.




















