Massive single-cell study by Kanai et al (www.medrxiv.org/content/10.1...):
- Once statistical power is high, constrained genes have more (though weaker) eQTLs.
- Chromatin-QTLs near constrained genes have "normal" effect sizes, colocalize more with disease, but exhibit attenuated peak-gene effects.
30.11.2025 17:06 — 👍 33 🔁 9 💬 1 📌 0
I haven't found it printed but there is a PDF link here: www.ashg.org/wp-content/u...
16.10.2025 21:17 — 👍 1 🔁 1 💬 0 📌 0

Image of an old building in Oxford with the heading 'postdoc opportunities' and the text 'computational approaches to improve rare disease diagnosis and treatment' and 'Big Data Institute, University of Oxford'
📣 We are recruiting! Please share!!
Are you a bioinformatician / computational scientist who wants to apply your skills to understanding regulatory biology and improving rare disease diagnosis and treatment? 🧠 💻 🧬 🩺
We have two roles available 👇
🧵 1/4
31.07.2025 16:12 — 👍 43 🔁 43 💬 1 📌 3
Isn't genetics cool???
Within only 145 nucleotides(!) of a non-coding RNA (RNU4-2) - different variants in distinct regions / structures cause three distinct disorders!!! (all discovered within the last 18 months)
🤯🤓🧬❤️
18.08.2025 14:03 — 👍 62 🔁 13 💬 1 📌 2

🗣️ Quote of #ESHG2025 (so far)
"Who licks bone !?!" 🦴
- Johannes Krause
Anyone have that on your bingo card?
Well apparently archeologists do, to distinguish bone from stones and it causes problems in DNA sequencing. 🤔
24.05.2025 13:46 — 👍 28 🔁 10 💬 1 📌 0
We are just wrapping up day 1 at #ESGH2025 in beautiful Milan. For those who want some extra fun while listening to the great science, you can play bingo.👇
I know multiple of these have already occurred.
24.05.2025 17:14 — 👍 5 🔁 0 💬 1 📌 0
Buongiorno Milano! Ready for a great day 1 of #eshg2025?
Packed program of excellent science 8.30am-8.00pm - plus networking event till 9.30pm to meet many friends, colleagues and collaborators! …andiamo @eshg.bsky.social @eshgyoung.bsky.social
24.05.2025 04:45 — 👍 15 🔁 6 💬 0 📌 0
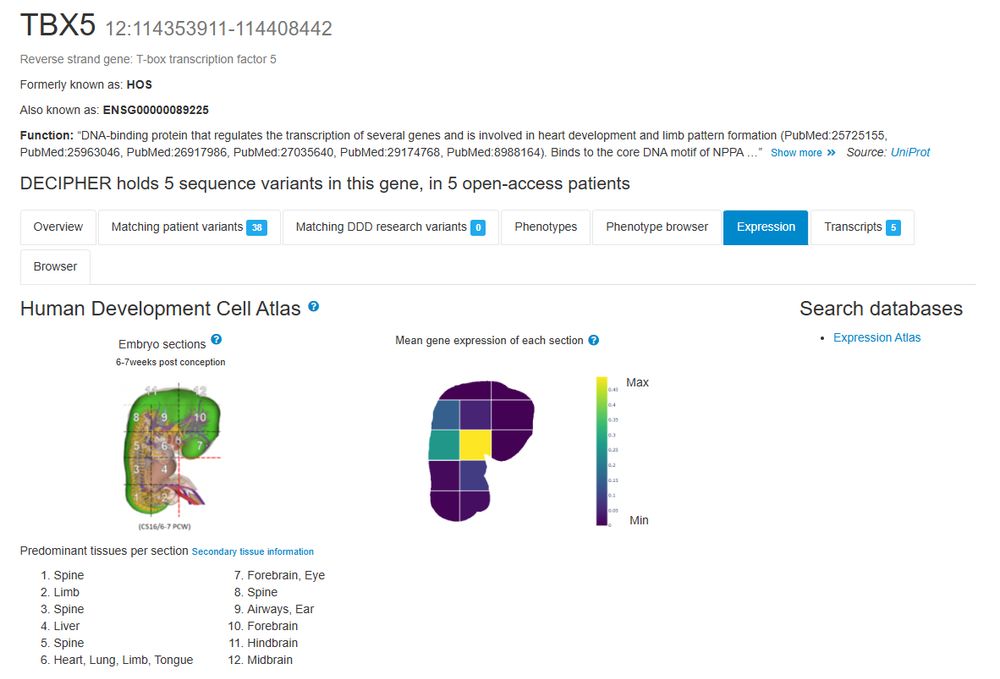
Human Developmental Cell Atlas (HDCA) expression data is now displayed. Expression is displayed in 12 sections of a 6-7 post-conception week human embryo, alongside a sagittal view which displays the region of the embryo represented by each section @mhaniffa.bsky.social
07.05.2025 13:29 — 👍 17 🔁 7 💬 0 📌 1

My son Charlie — and the breakthrough that changed our lives
James Coney and his wife, Sarah, struggled not knowing why their 12-year-old was born with a severe learning disability. In their darkest moments, they blamed themselves. Then, out of the blue, came a...
A few weeks ago, I had an incredibly emotional call with James Coney, a writer for the Sunday Times whose son Charlie was in the @genomicsengland.bsky.social 100k project and was recently diagnosed with ReNU syndrome. This beautiful article tells their story ❤️ www.thetimes.com/article/0bcc...
02.03.2025 12:06 — 👍 111 🔁 41 💬 5 📌 6
Yes, I've done this once before for a paper (listing in the system only first and last authors). It is easy to get those handful of people to approve additional authors in the future if the journal requires it.
Not sure if recommended, but it saved time.
21.02.2025 17:33 — 👍 1 🔁 0 💬 0 📌 0

Genomics of Rare Disease (hybrid conference)
Dates: 9-11 April 2025
Location: Hinxton Hall Conference Centre, Wellcome Genome Campus, UK and online
In-person registration deadline: 11 March 2025
Virtual registration deadline: 1 April 2025
Join leading experts working in #RareDisease research at our #GRD25 conference.
📅 Dates: 9-11 April 2025
💭 Share insights in person
Explore the latest #genomics advances accelerating improvements in clinical care for rare disorders, globally.
⏰Secure your place by 11 March: bit.ly/3BpAe44
17.02.2025 13:21 — 👍 9 🔁 12 💬 0 📌 6
Projects like this can’t be completed without many others: thanks to Mark Daly for continued mentorship across the years; critical support and work from @anneotation.bsky.social, @konradjk.bsky.social, Predrag Radivojac; the Hail team; and everyone associated with @gnomad-project.bsky.social.
10/10
19.04.2024 23:42 — 👍 1 🔁 0 💬 0 📌 0


If this work feels familiar, it is because it is building off older work from our team originally released for ExAC.
I view this iteration as more of a franchise reboot instead of a sequel – we have new leads, but similar themes.
9/10
19.04.2024 23:39 — 👍 1 🔁 0 💬 1 📌 0
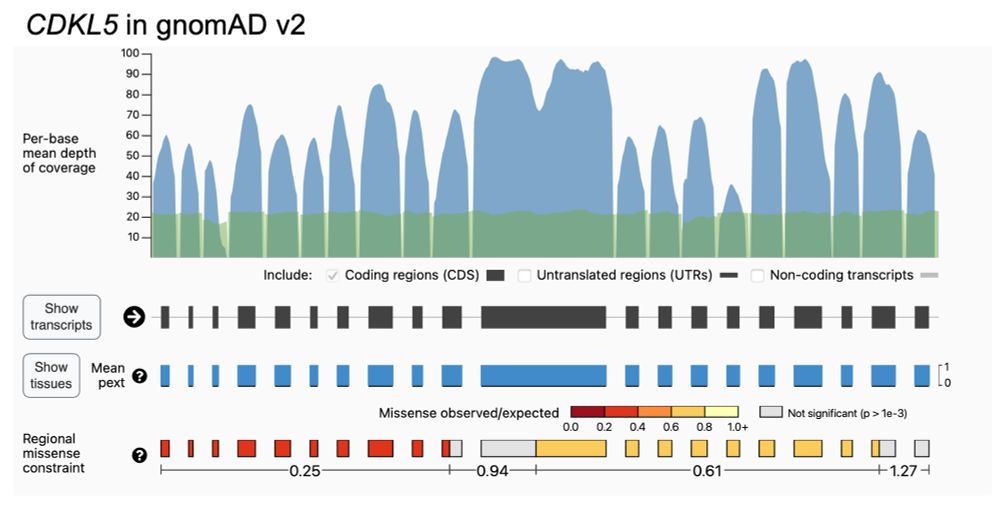
As with all gnomAD-led projects, we’ve already shared the data and code. Regions are displayed for v2 on the gnomAD browser, the code can be seen on Github (github.com/broadinstitu...), and MPC scores are available for download.
8/10
19.04.2024 23:39 — 👍 0 🔁 0 💬 1 📌 0
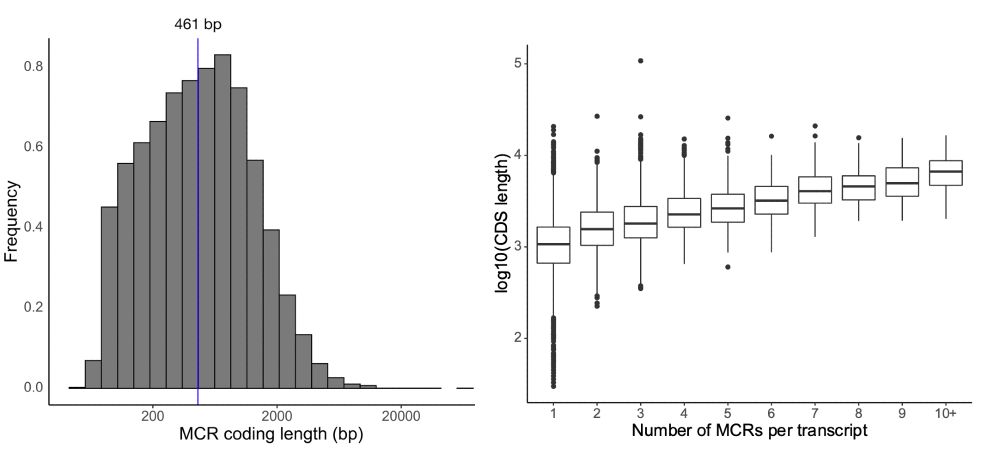
There is still much more to learn: using 125k exomes, our median region size is ~450bp and we see a relationship between transcript length and the number of regions we can identify due to statistical power.
7/10
19.04.2024 23:38 — 👍 0 🔁 0 💬 1 📌 0
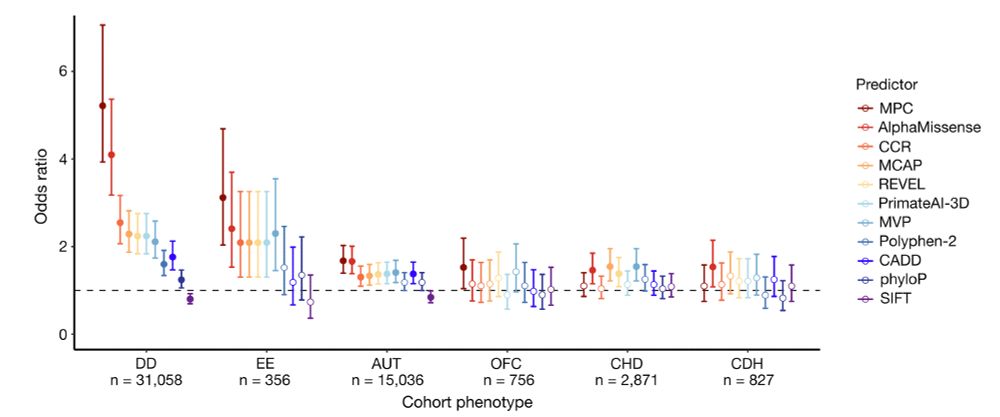
Finally, missense constraint information was incorporated into a deleteriousness metric named MPC (Missense deleteriousness Prediction by Constraint), which separates case from control de novo missense variants well with similar performance to ML models like AlphaMissense.
6/10
19.04.2024 23:37 — 👍 0 🔁 0 💬 1 📌 0

In collaboration with Predrag Radivojac and team, we demonstrated that coding bases with < 20% of their expected missense variation achieve moderate support for pathogenicity (PM1) following ACMG/AMP guidelines that can be used for clinical classification.
5/10
19.04.2024 23:37 — 👍 0 🔁 0 💬 1 📌 0
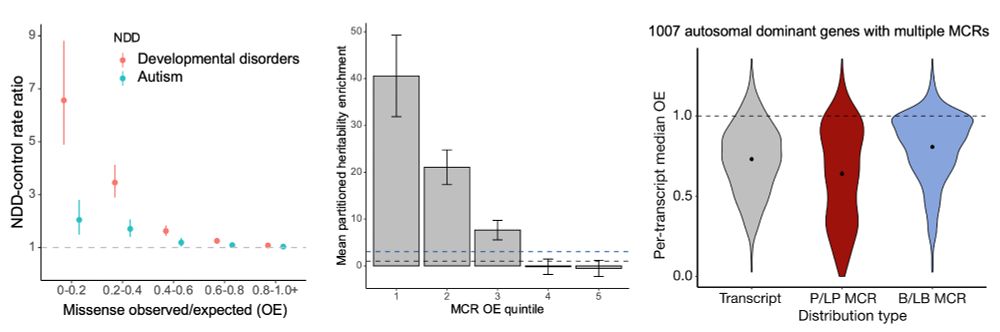
Missense depleted regions show an enrichment of (1) de novo missense variants in neurodevelopmental disorder cases compared to controls, (2) partitioned common variant heritability for >260 independent traits from the UK Biobank, and (3) ClinVar pathogenic (P/LP) variants.
4/10
19.04.2024 23:36 — 👍 0 🔁 0 💬 1 📌 0
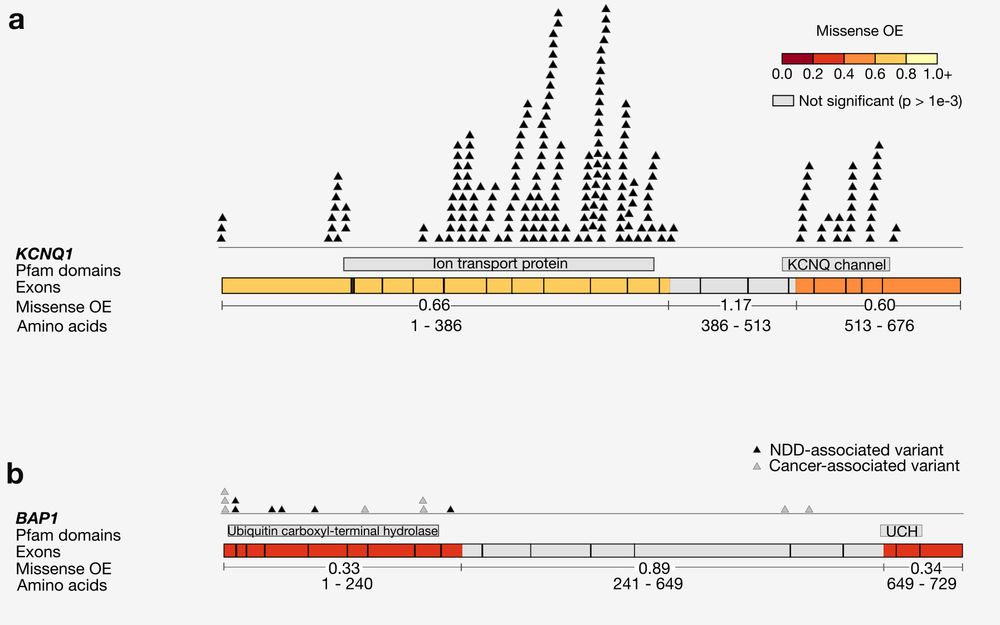
Why try to find subgenic regions that are specifically missense constrained? Splitting up genes reveals patterns of negative and neutral selection that are obscured when looking gene-wide, including highlighting regions that have a large number of known pathogenic variants.
3/10
19.04.2024 23:35 — 👍 0 🔁 0 💬 1 📌 0
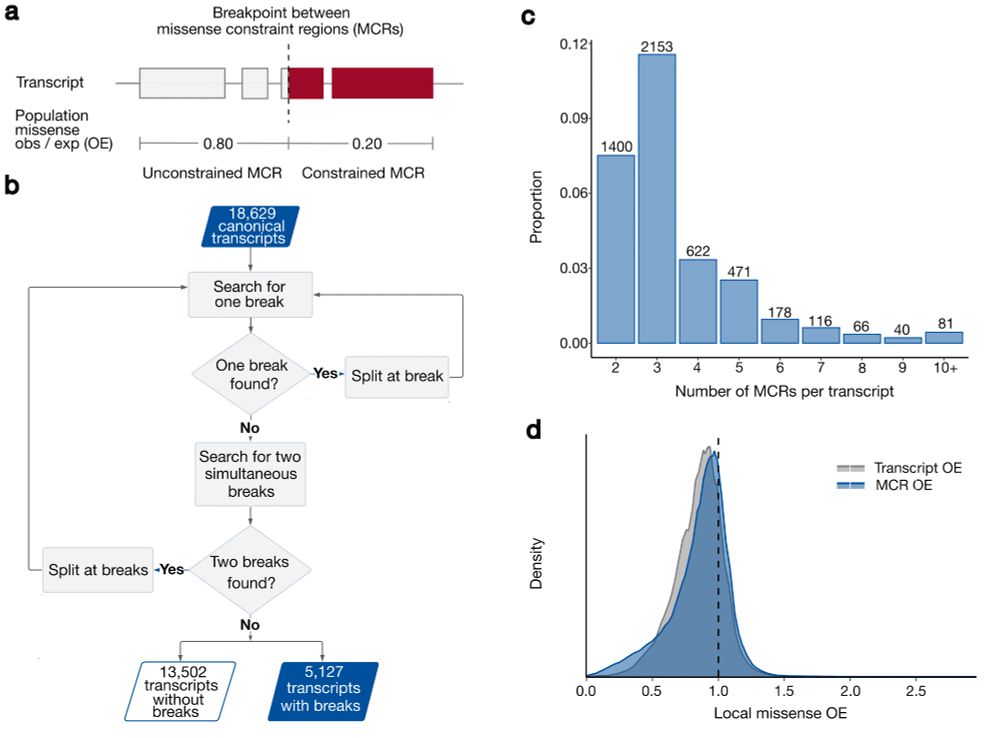
Co-led by the fabulous Katherine Chao and Lily Wang, we used gnomAD v2 and a recursive search to identify ~28% of canonical transcripts that were split into multiple missense constraint regions (measured by variable missense depletion in gnomAD).
2/10
19.04.2024 23:35 — 👍 0 🔁 0 💬 1 📌 0
Recently out on #bioRxiv: our updated approach to identify regional variability in missense mutation intolerance (“constraint”) in protein-coding genes using the gnomAD database.
www.biorxiv.org/content/10.1...
1/10
19.04.2024 23:34 — 👍 6 🔁 2 💬 1 📌 0
Some updated guidance on our gnomAD v4 constraint scores: gnomad.broadinstitute.org/news/2024-03...
The @gnomad-project.bsky.social team is hard at work on v4.1 and improvements across the board, so expect more updates.
Thanks to Katherine Chao for spearheading this blogpost.
08.03.2024 16:34 — 👍 2 🔁 1 💬 0 📌 0
A freely accessible version of the paper can be found here: rdcu.be/dsVXx
08.12.2023 02:38 — 👍 1 🔁 0 💬 0 📌 0
Our data are freely available on the gnomAD browser (gnomad.broadinstitute.org).
A huge thank you to Mike Guo, Laurent Francioli, Sarah Stenton, and Julia Goodrich for their hard work on this. This, of course, wouldn't be possible without everyone involved with @gnomad-project.bsky.social.
08.12.2023 02:38 — 👍 1 🔁 0 💬 1 📌 0
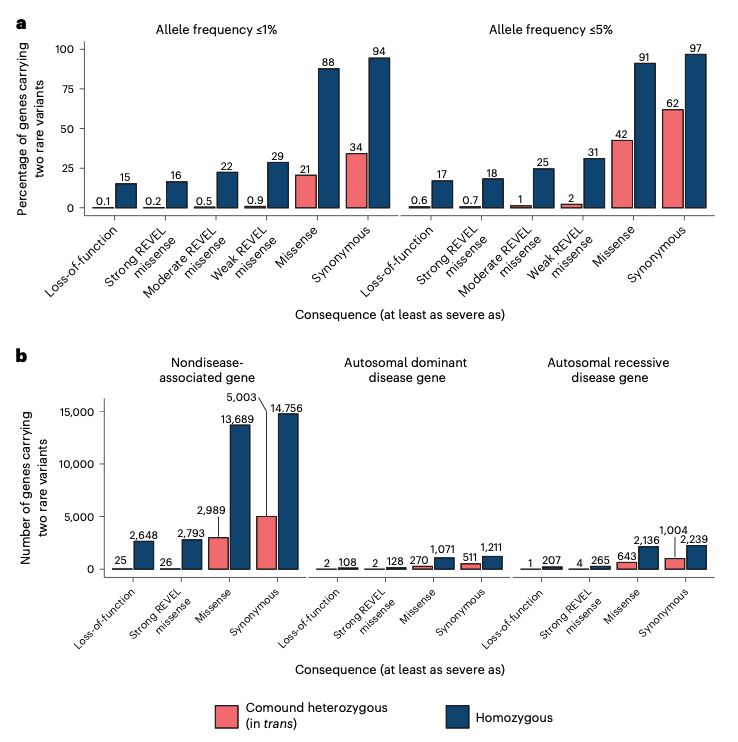
We also counted the number of genes that appeared to have compound heterozygous (in trans) deleterious variants and didn’t find that many. After manual curation, we only found seven genes with high-confidence compound heterozygous loss-of-function variants with some caveats.
08.12.2023 02:36 — 👍 2 🔁 0 💬 1 📌 0
Biomedical Science PhD student at UTHSC 🧬
In Dr. Mefford's lab @ St. Jude
Views are my own
Professor, Department of Medicine and McDonell Genome Institute @ Washington University. Specializing in Bioinformatics, Genomics, and Cancer. http://griffithlab.org/
Evolution, cancer, immunology, math(s)
Ursula Zoellner Professor of Cancer Research
University of Cambridge
blundelllab.com
Assistant Professor UTSW; Mutagenesis, Population genetics, Evolution in somatic tissues and on phylogenetic tree
Working to realize the promise of human genomics to better predict, prevent, and treat cancer. For more info: https://labs.dana-farber.org/collins-genomics/
Associate professor at the Hebrew University of Jerusalem.
Statistical, population, and medical genetics; preimplantation genetic testing. Views my own.
http://scarmilab.org
Professor. Sociologist. NYTimes Opinion Columnist. Books: THICK, LowerEd. Forthcoming: 1)Black Mothering & Daughtering and 2)Mama Bears.
Beliefs: C.R.E.A.M. + the internet ruined everything good + bring back shame.
“I’m just here so I don’t get fined.”
Assistant Professor of Genetics @ Yale.
Studying how variation in cis-regulatory-elements impacts evolution, complex traits, and more!
http://reilly-lab.com
Interdisciplinary scholar in psychiatry and mental health. My expertise is in schizophrenia, psychiatric genomics, and precision care—bridging research and practice to advance care outcomes
Professor of Psychology and Genetics at University of Surrey
Professor of Sociology at Purdue University. The Science Tooth Fairy.
Using social behavior and (epi)genetics to understand and predict depression.
Human disease genomics, precision medicine and machine learning. Associate Professor at @IcahnMountSinai
Group leader @metabolcenter.bsky.social and affil. with @broadinstitute.org Center for Genomic Mechanisms of Disease. Interested in brain control of metabolism using #hypothalamus #in-vivo/vitro/silico-models #singlecell #genetics #machinelearning.
Statistical Geneticist, Group leader at University of Lausanne/Unisante, father, climber, runner