Research Associate
Academic Job Category Faculty Non Bargaining Job Title Research Associate Department Wasserman Laboratory | Department of Medical Genetics | Faculty of Medicine (Wyeth Wasserman) Posting End Date Augu...
I'm hiring a Bioinformatics Research Associate for the Silent Genomes Project. PhD required, restricted to Canadians, work must be performed in British Columbia.
Great for those who love pipelines, whole genome data and work with a social purpose.
ubc.wd10.myworkdayjobs.com/ubcfacultyjo...
22.07.2025 20:42 — 👍 6 🔁 6 💬 0 📌 1

@saramostafavi.bsky.social (@Genentech) & I (@Stanford) r excited to announce co-advised postdoc positions for candidates with deep expertise in ML for bio (especially sequence to function models, causal perturbational models & single cell models). See details below. Pls RT 1/
19.06.2025 20:55 — 👍 55 🔁 40 💬 1 📌 3
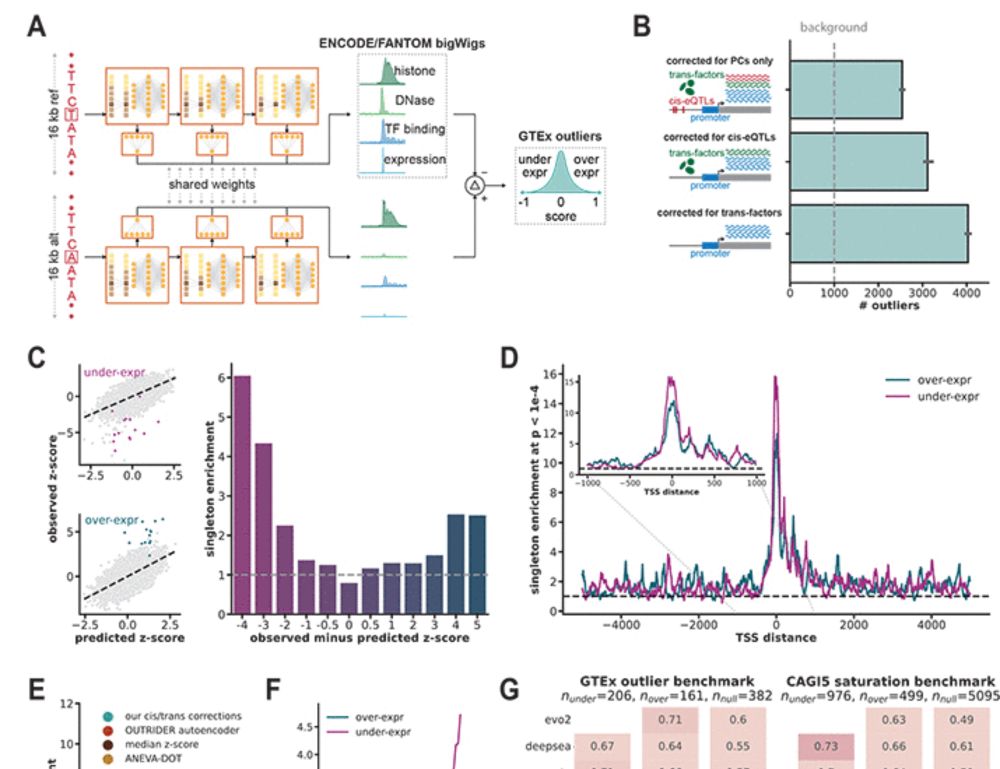
Yes, that's exactly what it is. Predicting the difference here is important.
10.06.2025 16:40 — 👍 1 🔁 0 💬 0 📌 0
This ensures the model focuses on the actual variant and doesn't overfit to correlated but irrelevant features, which leads to a better generalization.
10.06.2025 06:47 — 👍 1 🔁 0 💬 1 📌 0
Certainly! Here the entire model is shared, two copies see inputs that differ only at a single base pair (a variant of interest), and the model weights are tuned to learn the difference in effect size correctly.
10.06.2025 06:47 — 👍 1 🔁 0 💬 1 📌 0
Great question! That's our best guess as well and we highlight this in the paper by saying that MPRA experimental data from individual cell lines could have limitations for variant interpretation.
10.06.2025 06:46 — 👍 1 🔁 0 💬 0 📌 0
Huge thanks to the amazing Illumina team—this was an incredible learning experience! I'm excited to keep pushing forward as we develop models to tackle gene expression and non-coding variant interpretation. (16/)
29.05.2025 23:57 — 👍 2 🔁 0 💬 0 📌 0
A complementary thread from my colleague Kishore Jaganathan @kjaganatha.bsky.social bsky.app/profile/kjag... (15/)
29.05.2025 23:57 — 👍 2 🔁 0 💬 1 📌 0
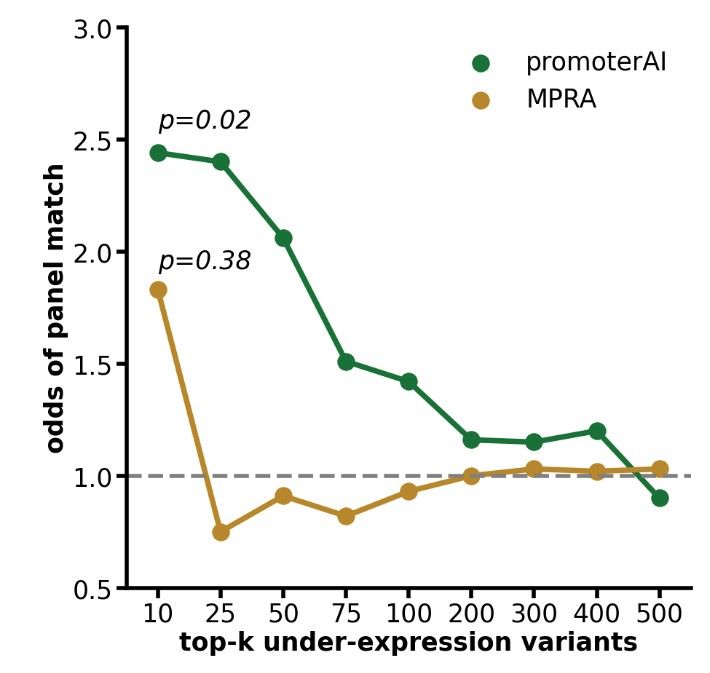
We followed up by testing promoter variants in Mendelian genes using MPRA. Surprisingly, PromoterAI was more effective than MPRA at prioritizing variants linked to patient phenotypes, highlighting limitations of MPRA for rare disease interpretation. (13/)
29.05.2025 23:57 — 👍 1 🔁 0 💬 2 📌 0
While we noticed that the use of additional species such as mouse does not lead to substantial improvement of variant effect prediction, it does help with ensembling. Thus, the final model is an ensemble of two: trained on human only and trained on mouse+human together. (12/)
29.05.2025 23:57 — 👍 1 🔁 0 💬 1 📌 0
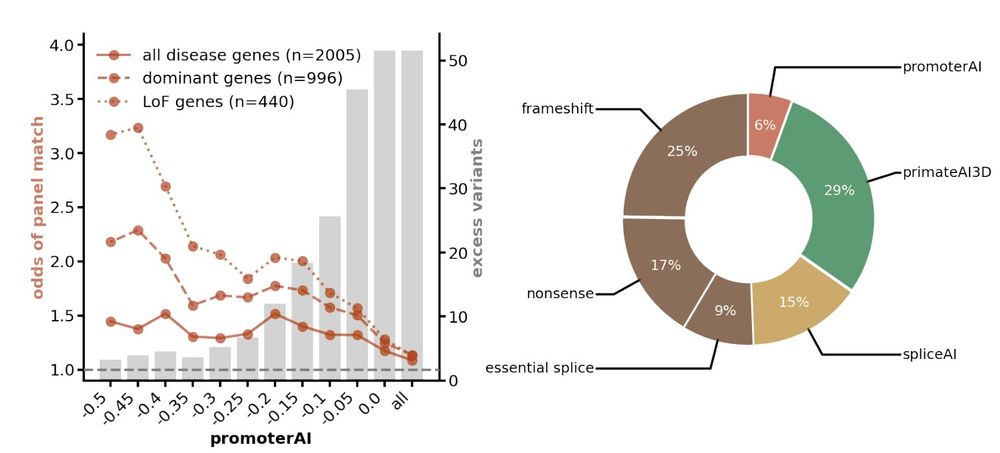
In the Genomics England rare disease cohort, functional promoter variants predicted by PromoterAI were enriched in phenotype-matched Mendelian genes. These variants accounted for an estimated 6% of the rare disease genetic burden. (11/)
29.05.2025 23:57 — 👍 2 🔁 0 💬 1 📌 0
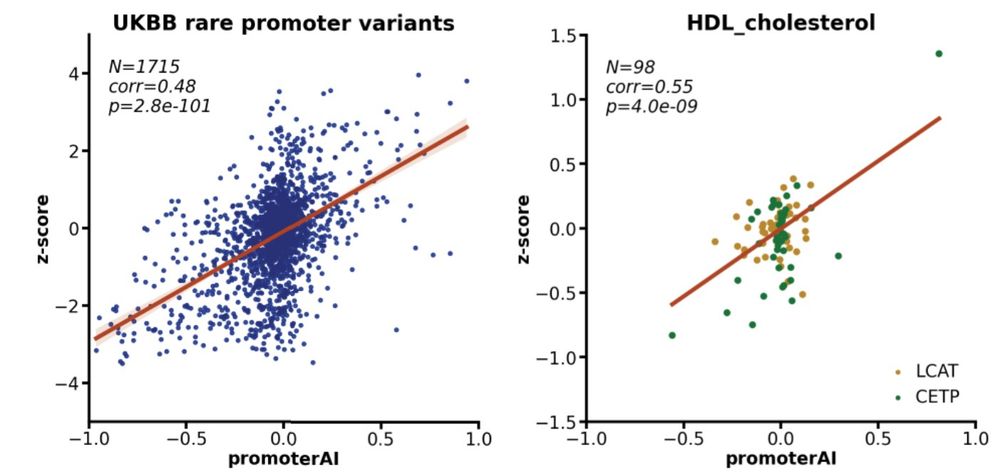
In the UK biobank cohort, PromoterAI's predicted promoter variant effects correlated strongly with measured protein levels and quantitative traits, suggesting that promoter variants contribute meaningfully to phenotypic variation in the general population. (10/)
29.05.2025 23:57 — 👍 1 🔁 0 💬 1 📌 0
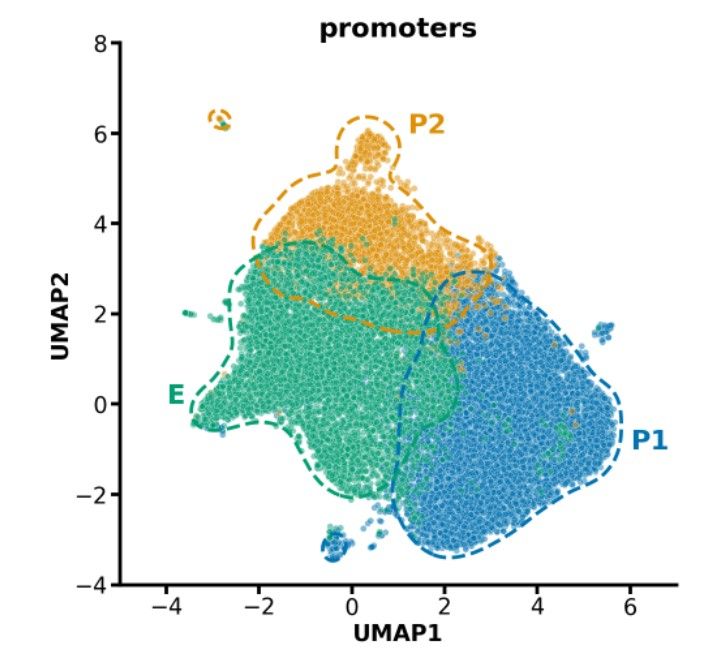
PromoterAI's embeddings split promoters into three distinct classes: P1 (~9K genes, ubiquitously active), P2 (~3K genes, bivalent chromatin), E (~6K genes, enhancer-like). The E class, enriched for TATA boxes, may reflect enhancers co-opted as promoters. (9/)
29.05.2025 23:57 — 👍 1 🔁 0 💬 1 📌 0
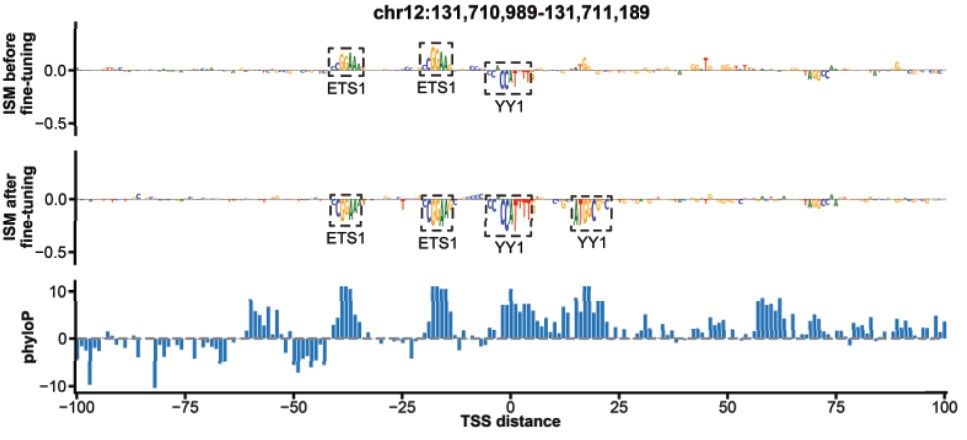
Fine-tuning improved PromoterAI’s ability to predict the direction of motif effects — a known issue of multitask models. The model often recognized motifs before fine-tuning, but got the direction wrong. After fine-tuning, its predictions aligned better with the data. (8/)
29.05.2025 23:57 — 👍 2 🔁 1 💬 1 📌 0
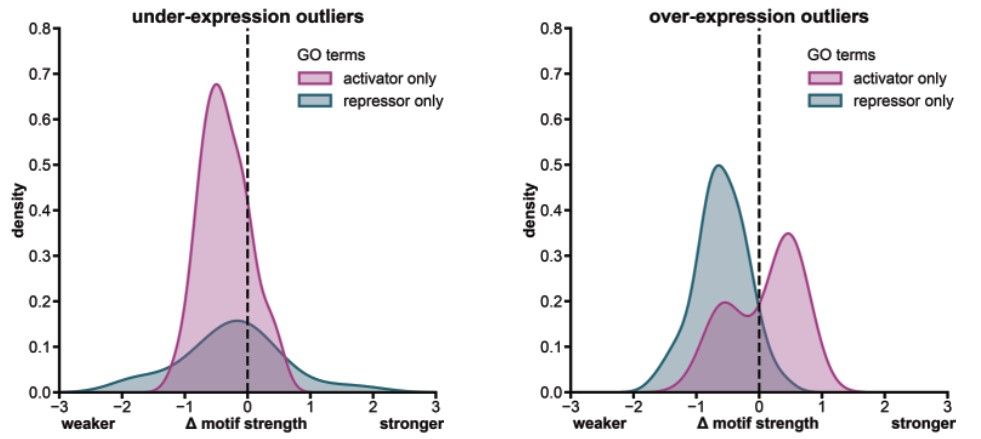
We used our list of gene expression outliers to explore their effect on transcription factor binding sites. Our results show that it is easier for new variants to cause outlier gene expression by disrupting existing regulatory components rather than creating new ones. (7/)
29.05.2025 23:57 — 👍 1 🔁 0 💬 1 📌 0
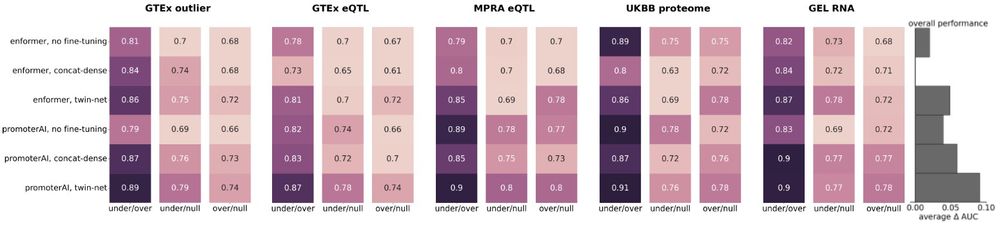
We also attempted to fine-tune Enformer and Borzoi on our promoter variant set. While performance improved, both models lagged behind PromoterAI. Notably, PromoterAI outperformed Enformer and was similar to Borzoi before fine-tuning. (6/)
29.05.2025 23:57 — 👍 4 🔁 1 💬 1 📌 0
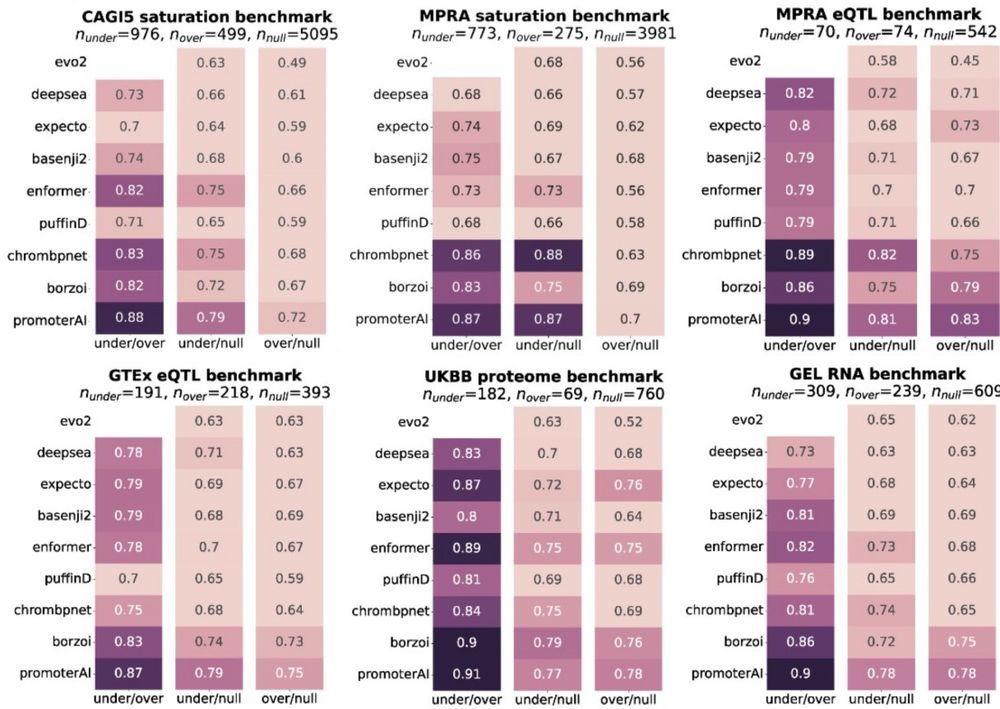
When it comes to predicting expression effects of promoter variants, PromoterAI achieved best performance across benchmarks spanning RNA, proteins, QTLs, and MPRA. (5/)
29.05.2025 23:57 — 👍 1 🔁 0 💬 1 📌 0
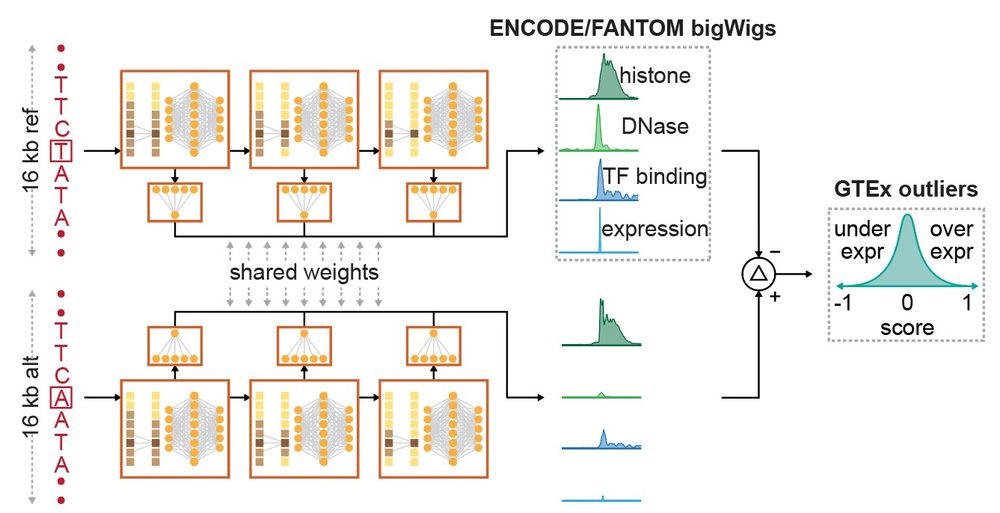
The second step was to fine-tune the model using a carefully curated list of rare promoter variants linked to aberrant gene expression. The fine-tuning was done using a twin-network setup to ensure the generalization across unseen genes and datasets. (4/)
29.05.2025 23:57 — 👍 3 🔁 0 💬 2 📌 0
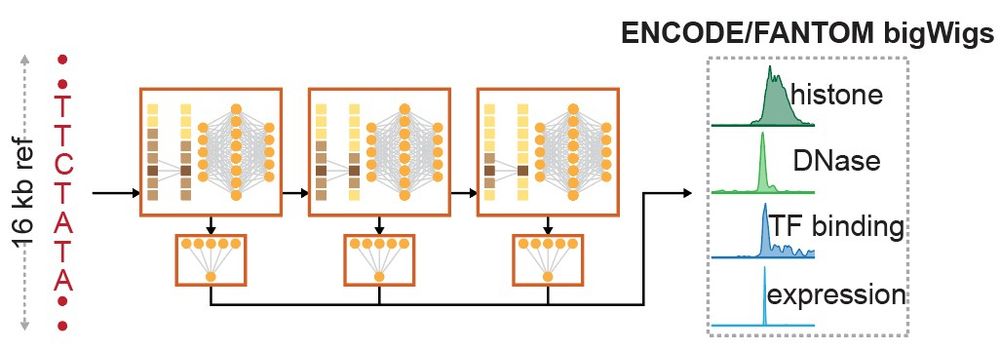
First, we pre-trained PromoterAI to predict histone marks, TF binding, DNA accessibility, and CAGE signal from a genomic sequence. The key difference with models like Enformer and Borzoi is that we predict at a single base-pair resolution and use only TSS-centered regions. (3/)
29.05.2025 23:57 — 👍 2 🔁 0 💬 1 📌 0
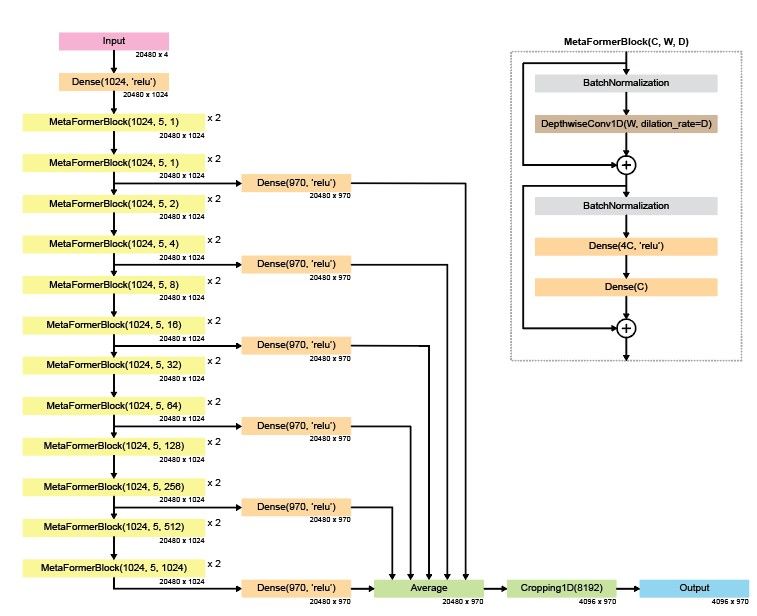
PromoterAI is built from transformer-inspired blocks called metaformers — but instead of attention, we use depthwise convolutions, making it a fully convolutional model. We believe that CNN-based methods are not surpassed yet and remain a great choice for genomics tasks. (2/)
29.05.2025 23:57 — 👍 3 🔁 0 💬 1 📌 0
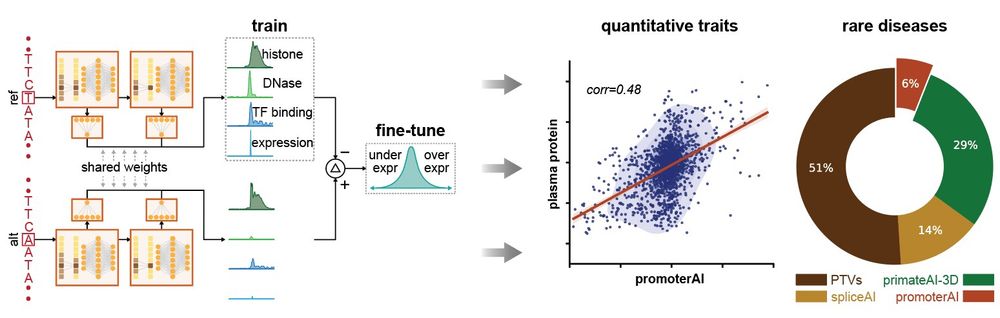
Excited to share my first contribution here at Illumina! We developed PromoterAI, a deep neural network that accurately identifies non-coding promoter variants that disrupt gene expression.🧵 (1/)
29.05.2025 23:57 — 👍 59 🔁 21 💬 1 📌 1
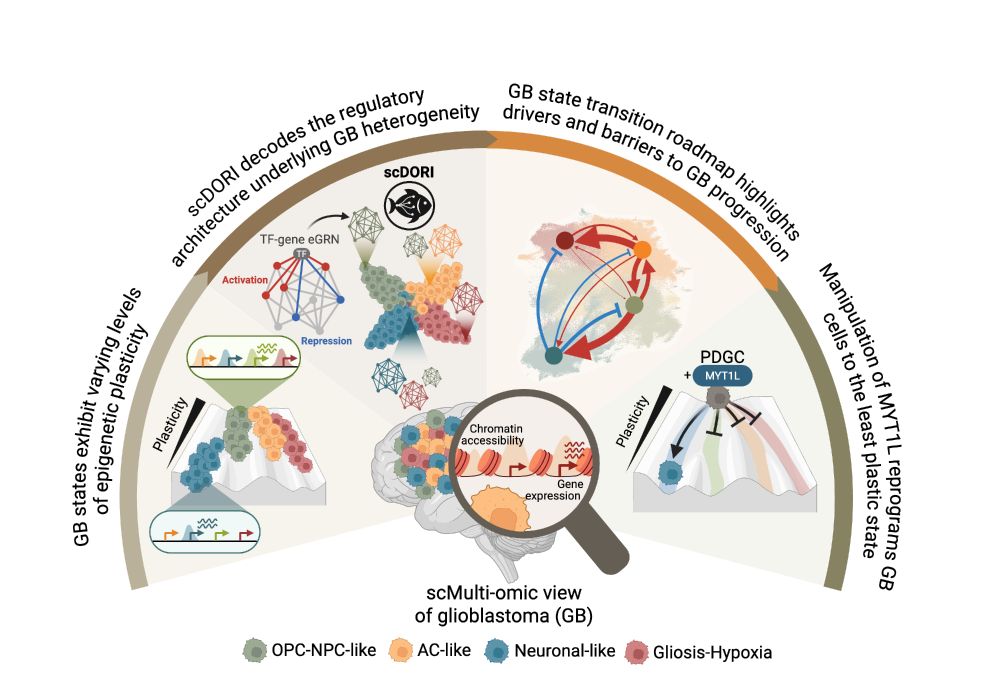
🧠 Excited to share my main PhD project! We mapped the regulatory rules governing Glioblastoma plasticity using single-cell multi-omics and deep learning. This work is part of a two-paper series with @bayraktarlab.bsky.social @oliverstegle.bsky.social and @moritzmall.bsky.social, Preprint at end🧵👇
16.05.2025 10:04 — 👍 76 🔁 29 💬 1 📌 6
So excited to see Yawei's manuscript on long-range MPRAs out! Some really great insights into distal enhancer regulation 🙂
23.04.2025 21:10 — 👍 15 🔁 4 💬 1 📌 0

Day 1 of #RECOMB2025-SEQ starts with @rayanchikhi.bsky.social and an introduction to Logan, a planetary-scale effort to assemble everything.
Currently cataloguing about 700k virus species!
📄 www.biorxiv.org/content/10.1...
24.04.2025 01:12 — 👍 20 🔁 4 💬 1 📌 0
Our latest work now online in Cell:
Rewriting regulatory DNA to dissect and reprogram gene expression
Our new method (Variant-EFFECTS) uses high-throughput prime editing + flow sorting + sequencing to precisely measure effects of noncoding variants on gene expression
Thread 👇
17.04.2025 18:26 — 👍 117 🔁 28 💬 1 📌 2
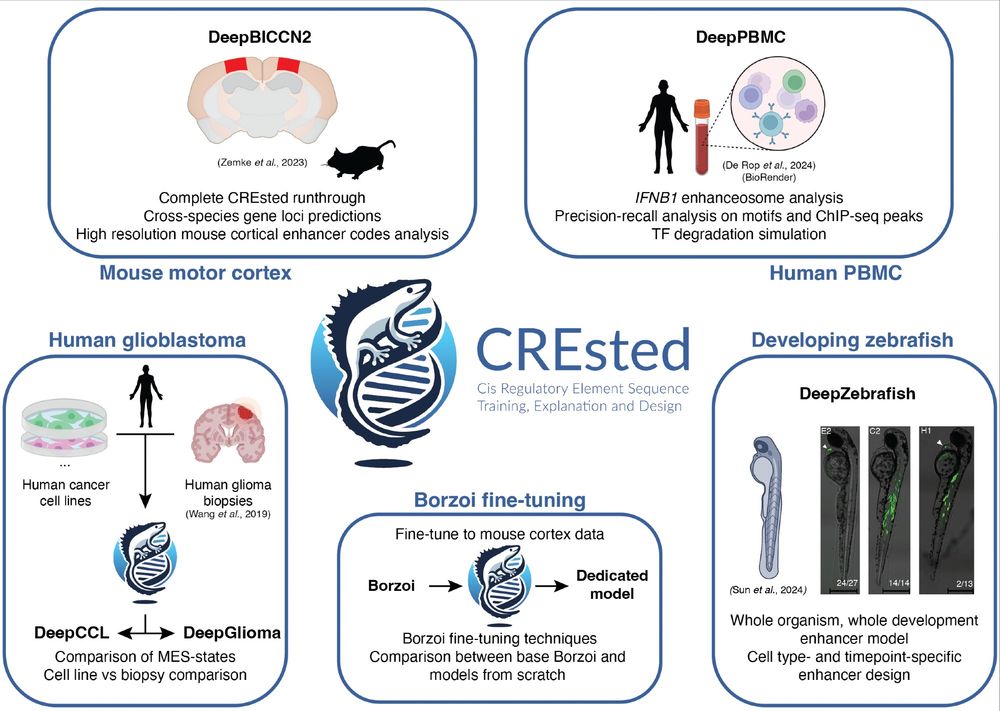
We released our preprint on the CREsted package. CREsted allows for complete modeling of cell type-specific enhancer codes from scATAC-seq data. We demonstrate CREsted’s robust functionality in various species and tissues, and in vivo validate our findings: www.biorxiv.org/content/10.1...
03.04.2025 14:30 — 👍 75 🔁 38 💬 1 📌 5
PhD student in Bioinformatics at the University of British Columbia
Data science, single cell sequencing, genome regulation, stem cell differentiation
CS PhD student @ Berkeley | https://rastogiruchir.github.io/
Genomics biologist. Group leader at MRC Laboratory of Medical Sciences, Hon. Senior Lecturer (Associate Prof.) at Imperial College London.
Opinions / view my own.
https://functionalgenecontrol.group
PhD student @uwcse; ML&CompGenomics&GeneRegulation; Bio&CS undergrad
@PKU1898; Intern@Yale, Microsoft Research Asia, Genentech
Statistician and computational biologist; uw alum; jhu student. He/him. http://ekernf01.github.io
dad, lab head, brewer, baker, beta cell maker
https://lynnlab.com
Genomics | Science | Technology | Biotech | Nonsense | All views are my own and not those of my employer- who is not on 🦋 yet.
Particular soft spot for single cell and spatial transcriptomics and genome biology.
PhD candidate in ML for genomics in Heidelberg, Germany with Oli Stegle
Previously at Genentech, UBC and BITS Pilani
https://scholar.google.com/citations?user=4yUtALcAAAAJ&hl=en&oi=ao
Postdoc studying chromatin regulation of behavior in the Deisseroth lab. PhD w/ Gerald Crabtree and former Allis lab member.
Stadtman Principal Investigator @NIH / NIA. Computational Genomics, RNA, Aging, Stress Response, Senescence. Posts are my own.
AI, BioML, compbio, data science | pathogens, AMR antimicrobial and antibiotic res, inflammation, CBD, single-cell RNA seq, metagenomics | Asst Prof @ University of Florida, AI advisor @ enGenome | Prev: UMich, UniPV, KyotoU | Immigrant 🇪🇺🇭🇰🇯🇵🇺🇸
Genomics, AI, sequence-to-function models, mechanisms of the cis-regulatory code. Investigator at the Stowers Institute.
Genetics, bioinformatics, comp bio, statistics, data science, open source, open science!
Scientist at Champalimaud Foundation (polaviejalab.org) and co-founder of Algebraic AI (algebraic.ai). #Maths4AI #AI4Science, #BehaviorAI #NeuroAI #CognitionAI #CollectiveBehavior
Computational biologist @HelmholtzMunich, prof @TU_Muenchen & associate PI @sangerinstitute. Dad of 4 and mountain lover. Department news, see @CompHealthMuc
Informaticist, Health Scientist & Developer. Building applications and systems. Just trying to help humanity + enjoy life. Need to try harder, probably.
Founder @informatics.fyi | Building @cambigo.com