Kudos to Sonia Zumalave in my lab for working out how to flag and remove such fold-back-like artifacts, with key contributions from @hbelrick.bsky.social @carolinmsa.bsky.social and @jevalleinclan.bsky.social. Thread on our algorithm bsky.app/profile/isid... and ..
20.07.2025 23:35 — 👍 1 🔁 0 💬 1 📌 0
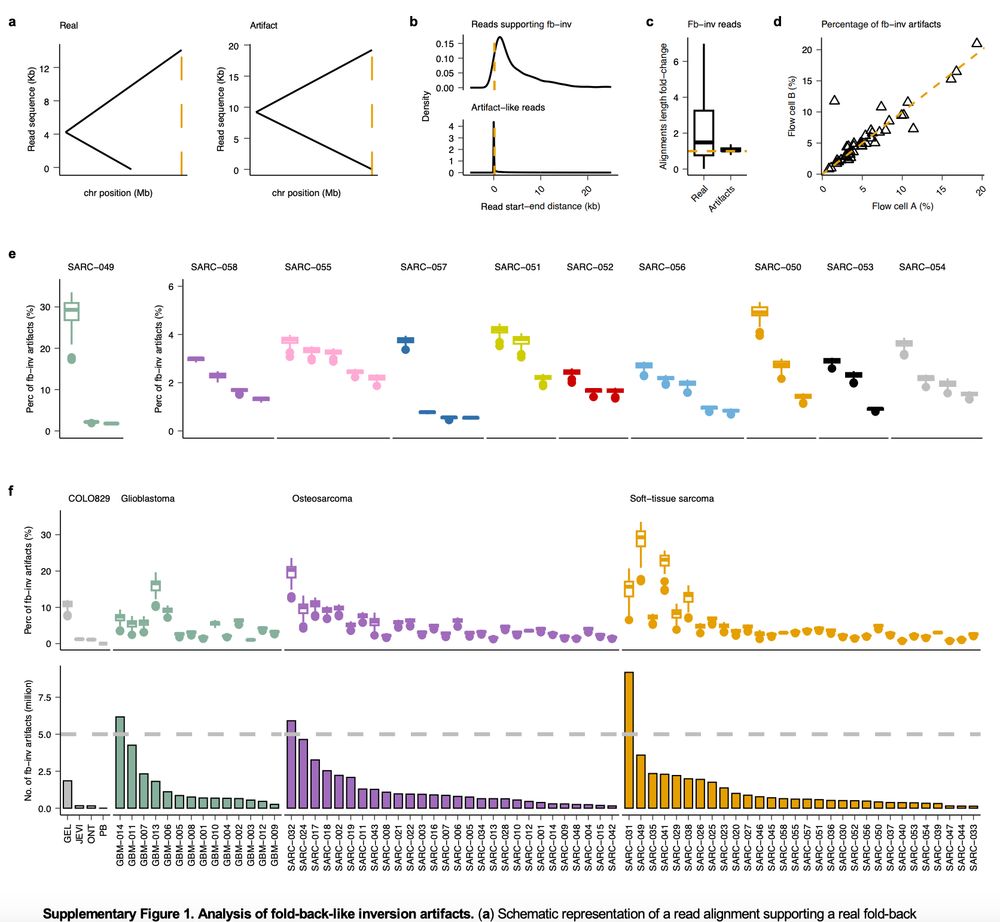
Very glad to see this preprint by @lh3lh3.bsky.social and Meyerson labs www.biorxiv.org/content/10.1... confirming our finding of artifactual fold-back inv in long reads (Fig S1 in our @natmethods.nature.com paper presenting SAVANA, which filters such artifacts to improve SV calling 👇
20.07.2025 23:35 — 👍 4 🔁 1 💬 1 📌 0
Work by researchers in the group of @isidrolauscher.bsky.social at EMBL-EBI, the R&D lab of @genomicsengland.bsky.social, in collaboration with clinical partners at @ucl.ac.uk, Royal National Orthopaedic Hospital, Instituto de Medicina Molecular João Lobo Antunes, and Boston Children’s Hospital.
29.05.2025 08:57 — 👍 3 🔁 1 💬 1 📌 0
Very well done indeed @hbelrick.bsky.social ! 😀
30.05.2025 12:41 — 👍 0 🔁 0 💬 0 📌 0
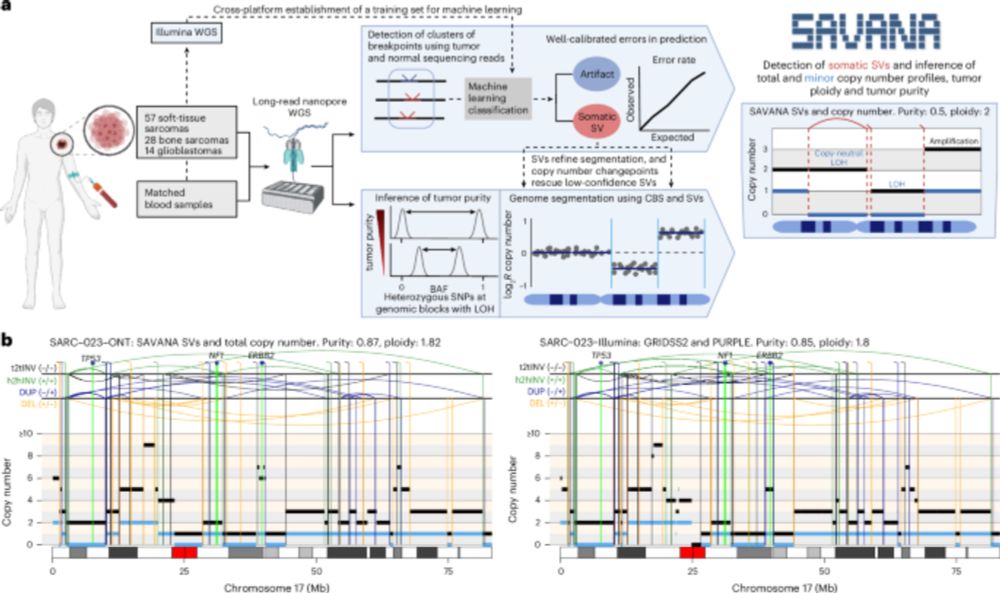
SAVANA is out in the wild 🦁! #SAVANA detects haplotype-resolved somatic structural variants (SVs), copy number aberrations, and calculates tumour purity and ploidy using long-read data. Together with it, a robust, data-driven benchmarking effort! Below is a thread with all the advantages 👇
29.05.2025 19:08 — 👍 7 🔁 3 💬 0 📌 0
and huge thanks to our funders 🙏 @curesarcoma.bsky.social CTOS, @embl.org and others!
28.05.2025 17:36 — 👍 0 🔁 0 💬 1 📌 0
Bluesky
HT @jevalleinclan.bsky.social nclan.bsky.social, and other lab members at @ebi.embl.org bl.org, our great collaborators at @bostonchildrens.bsky.social s.bsky.social Melanie Tanguy and Greg Elgar @genomicsengland.bsky.social sengland.bsky.social IMM Lisbon...
28.05.2025 17:36 — 👍 0 🔁 0 💬 1 📌 0
SAVANA was developed by two superstars in the lab @hbelrick.bsky.social & Carolin Sauer in close collaboration once again with Prof. Flanagan and team at @ucl.ac.uk with key contributions from...
28.05.2025 17:36 — 👍 0 🔁 0 💬 1 📌 0
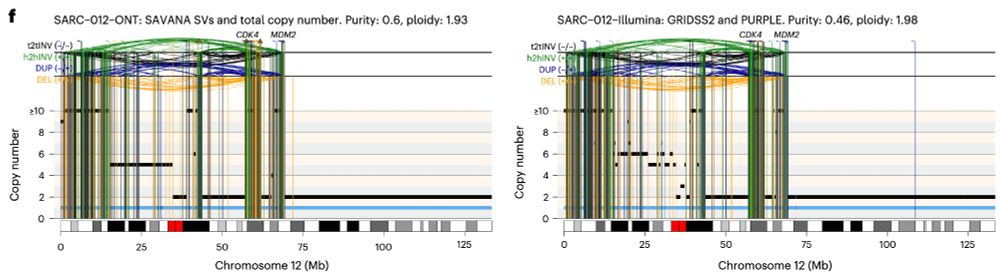
In sum, we establish best practices for benchmarking SV detection methods for somatic (eg cancer) genome analysis, and show that SAVANA enables the application of long-read sequencing to detect SVs and SCNAs reliably in clinical samples.
28.05.2025 17:36 — 👍 0 🔁 0 💬 1 📌 0
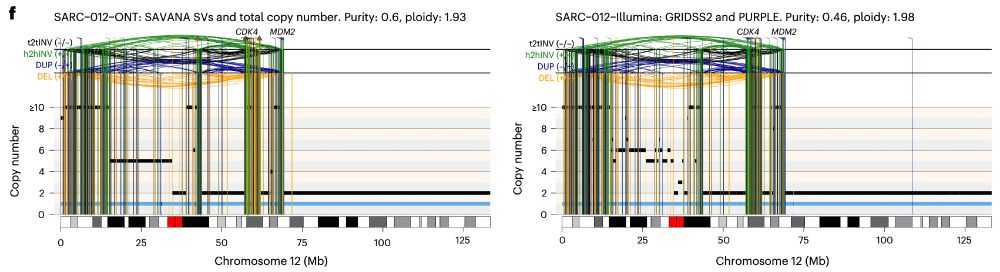
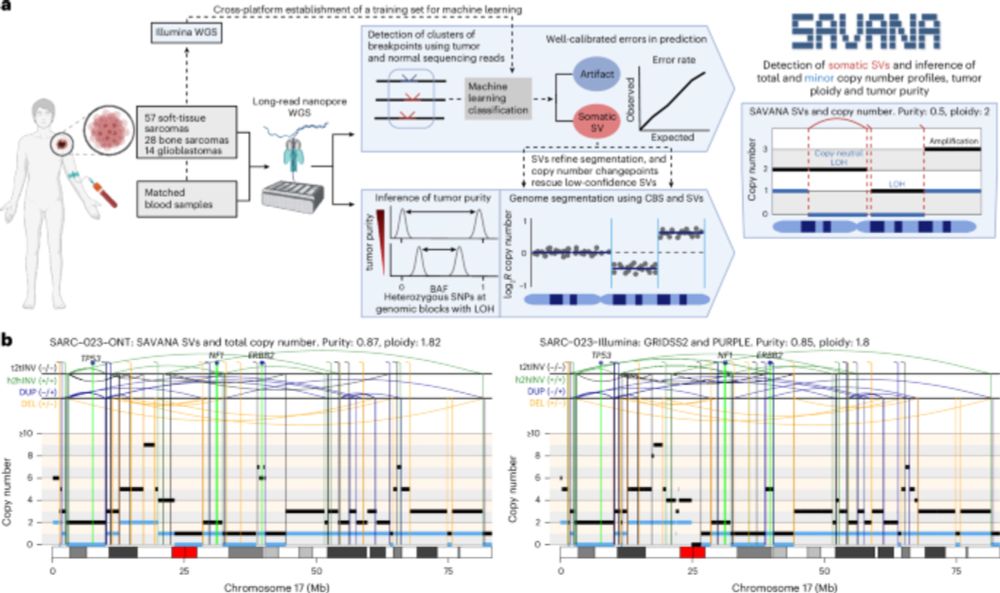
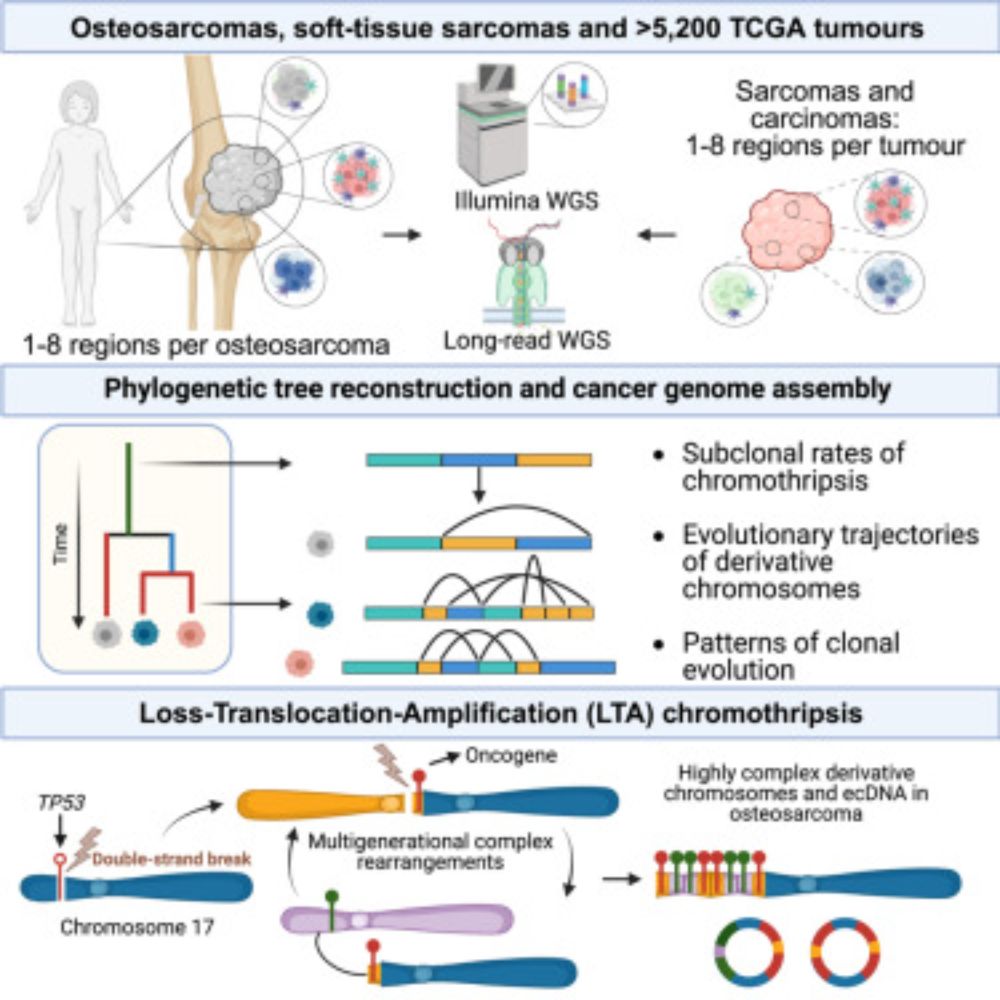
In practice, this means that we can now study, reliably, complex genomic rearrangements (e.g. #chromothripsis) and clinically relevant events causing tumour suppressor gene loss using long reads (left) with comparable accuracy to Illumina (right):
28.05.2025 17:36 — 👍 0 🔁 0 💬 1 📌 0
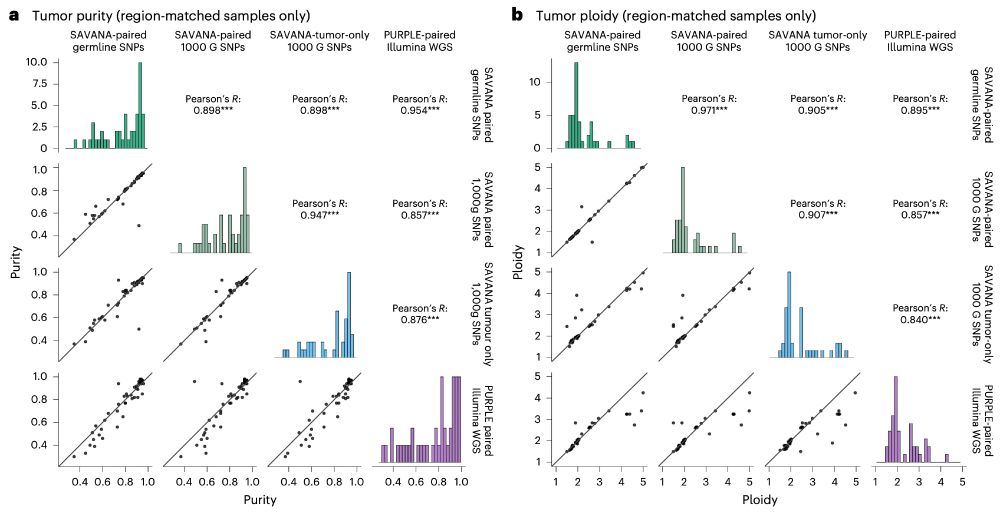
Moreover, using #SAVANA, we can estimate tumour purity and ploidy with comparable accuracy to illumina data (using the fantastic pipeline developed by the Hartwig Medical Foundation @ecuppen.bsky.social @danielisskeptical.bsky.social for clinical reports) even WITHOUT a germline control!
28.05.2025 17:36 — 👍 0 🔁 0 💬 1 📌 0
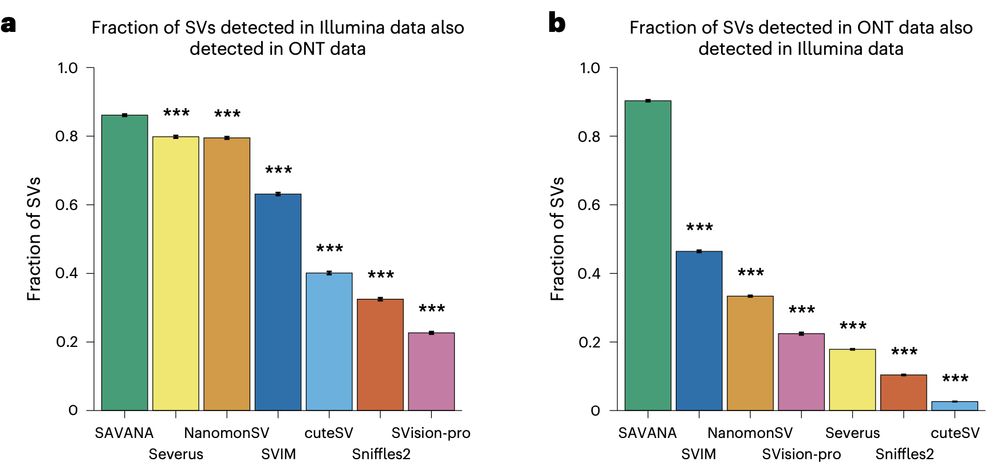
Using SAVANA, we recover most of the SVs detected in short-read data (note the higher than two-fold diff in coverage between long and short reads here!!), and most of the SVs detected using long reads are detected in illumina data (note that we are not using ultra-long reads)
28.05.2025 17:36 — 👍 0 🔁 0 💬 2 📌 0
Now that we have a robustly-validated algorithm we can address the question you are all waiting for (and which many colleagues have asked us many times): what is the relative performance of long & short reads to analyze human cancer genomes?
28.05.2025 17:36 — 👍 0 🔁 0 💬 1 📌 0
What underpins the higher performance of SAVANA? A key innovation of SAVANA is the use of machine learning to distinguish true somatic signal from artefacts. The key challenge here was to curate a large training set (see details in the paper).
28.05.2025 17:36 — 👍 0 🔁 0 💬 1 📌 0
In sum, these data indicate that SAVANA delivers SV results consistent with tumour biology, and the differences in SV rates across algorithms are caused by variable algorithmic performance, rather than true biological signal (see other analysis in support of this conclusion in the paper)
28.05.2025 17:36 — 👍 0 🔁 0 💬 1 📌 0
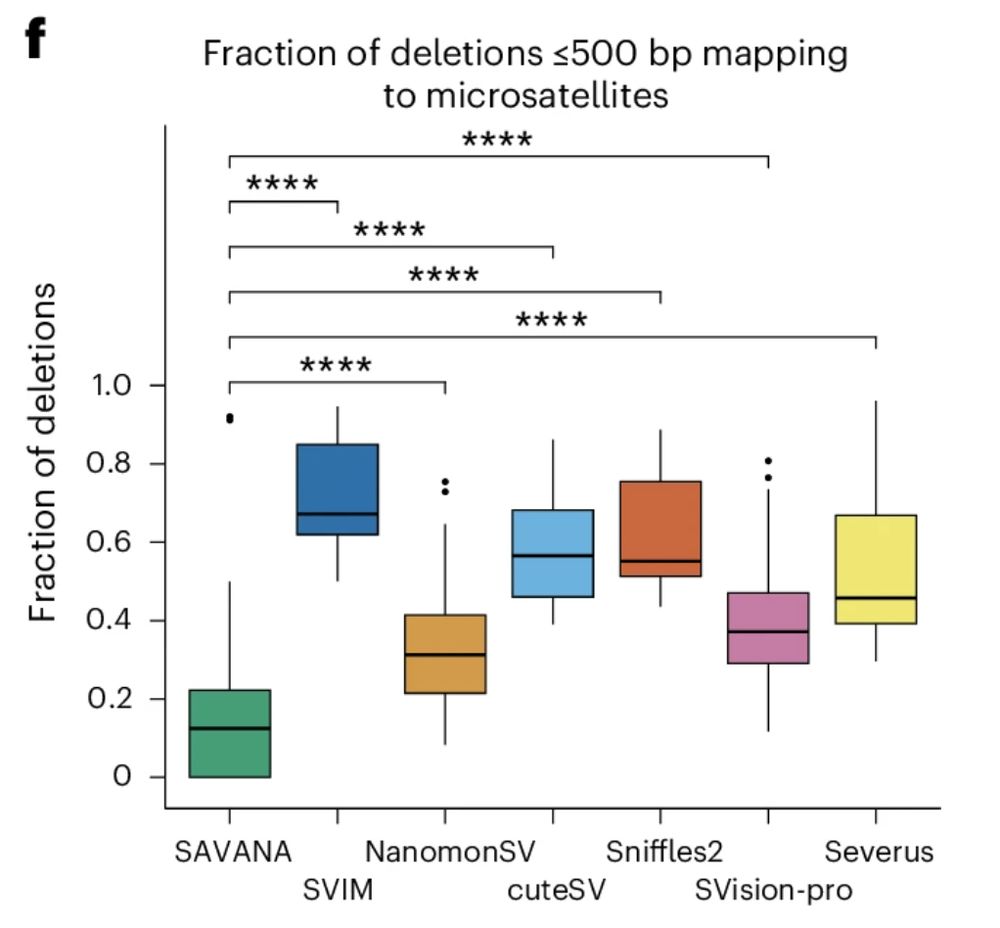
For example, existing methods detect 100s to 1000s of SVs in each sample mapping to microsatellite regions (#SAVANA doesn’t). The tumour types we analysed (sarcomas and glioblastomas) rarely show such levels of repeat instability, which we confirmed for our sample using illumina
28.05.2025 17:36 — 👍 0 🔁 0 💬 1 📌 0
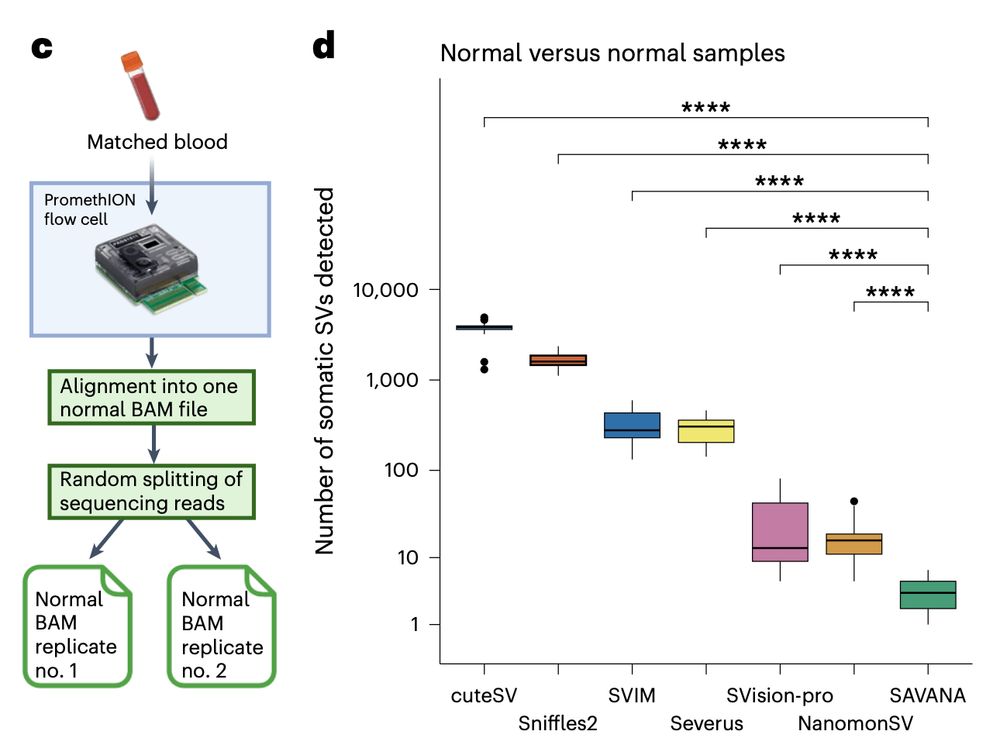
We found the same when using simulated sequencing replicates of the blood samples we use as germline controls. So, what are the false positive SVs called by some algorithms and not by others? What drives such strong differences in performance?
28.05.2025 17:36 — 👍 1 🔁 0 💬 1 📌 0
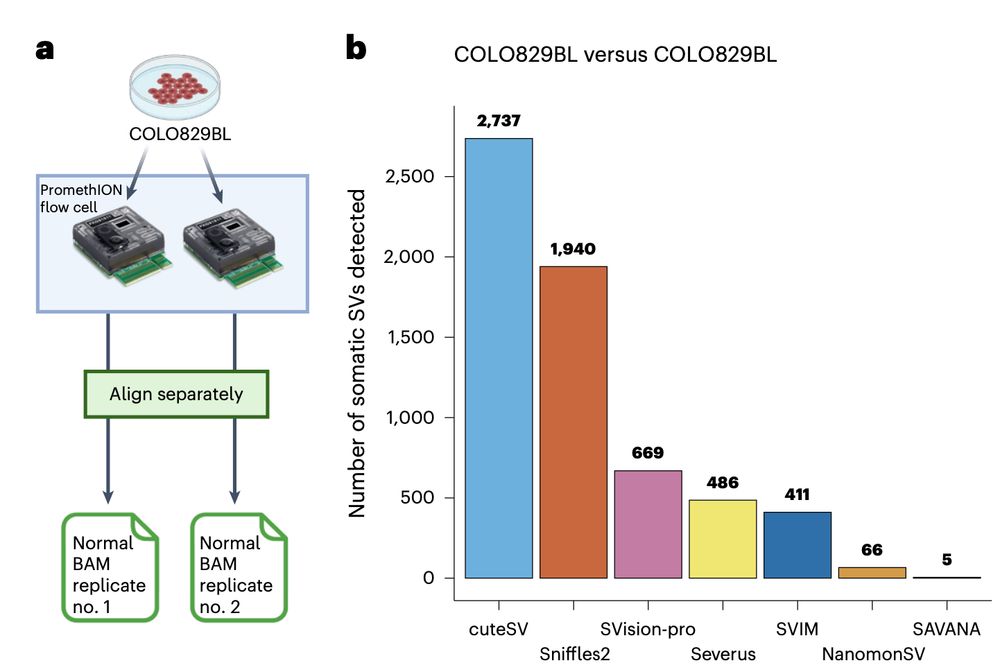
Using sequencing replicates of the normal cell line COLO829BL, we found that SAVANA shows 13- and 82-times higher specificity than the second and third-best performing algorithms (391x higher than the worse performing one). In practice, this means 10s-1000s less false positives..
28.05.2025 17:36 — 👍 1 🔁 1 💬 1 📌 0
Still not convinced? We also reasoned the following. If you use the same sample as both the tumour and matched germline sample to look for somatic SVs, how many would you detect? The answer is: 0, as you are comparing the same sample against itself. In other words: 1-1=0
28.05.2025 17:36 — 👍 0 🔁 0 💬 1 📌 0
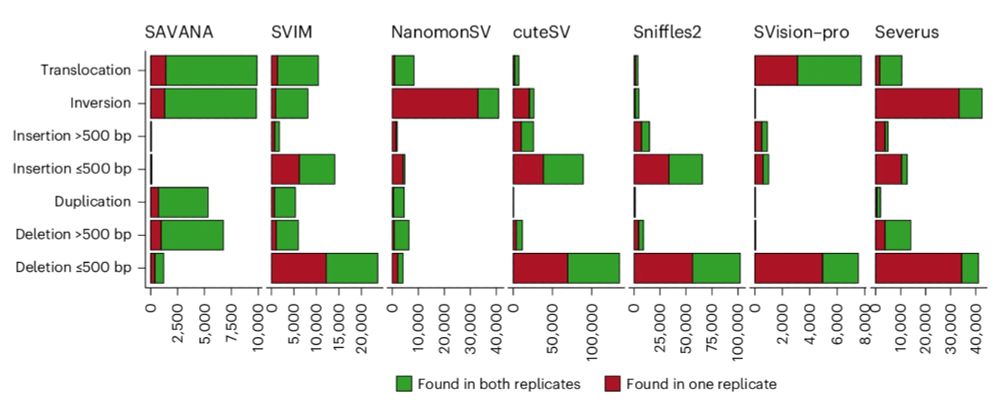
Well, nullius in verba, let the data speak. SAVANA shows uniform and much higher replication rates across SV clonality levels, sizes, types, samples and genomic regions. Thus, SAVANA's performance if driven by higher sensitivity and specificity!
28.05.2025 17:36 — 👍 0 🔁 0 💬 1 📌 0
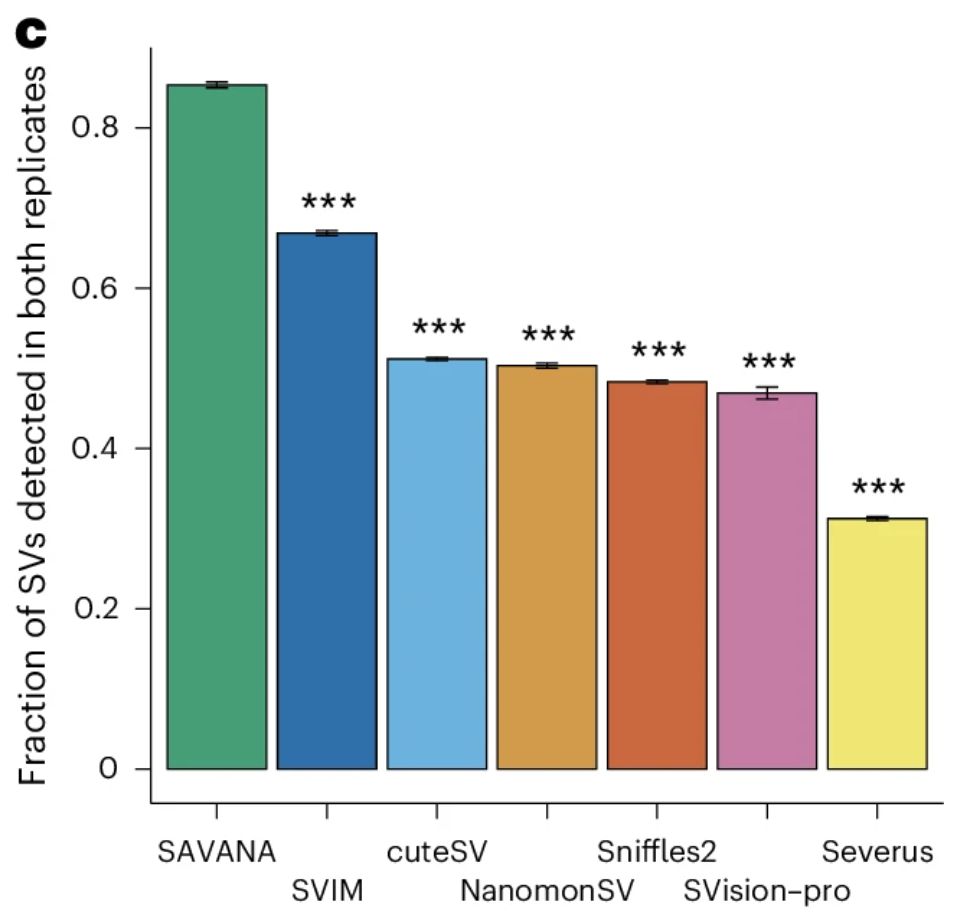
The replication rate of #SAVANA was much higher as compared to existing methods 👇One could argue that lowAF SVs might be missed in one replicate if reads supporting them are assigned to one replicate, and that the many SVs detected by some tools is due to higher sensitivity.
28.05.2025 17:36 — 👍 0 🔁 0 💬 1 📌 0
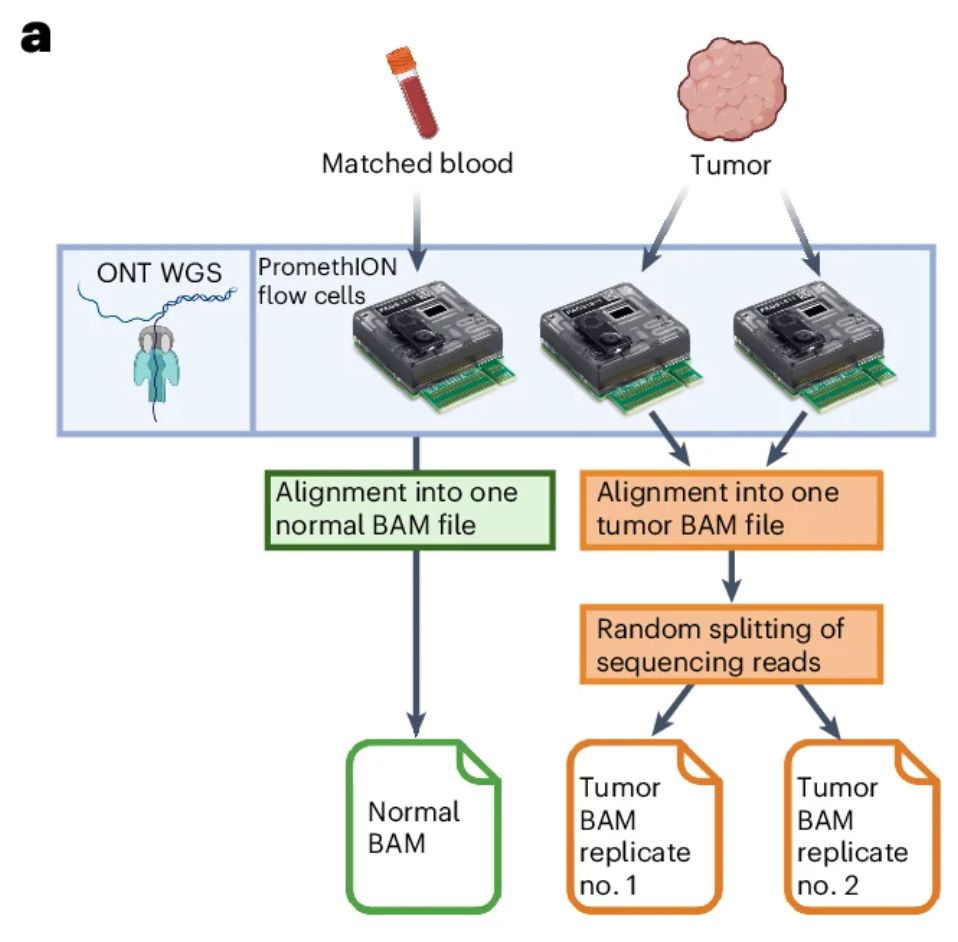
First, we simulated replicates in silico by randomly splitting the sequencing reads from each tumour, reaching even coverage per replicate (note that flowcell yield is variable for ONT). The key idea is: true SVs are detected in both replicates, false positives in just one
28.05.2025 17:36 — 👍 0 🔁 0 💬 1 📌 0
We propose to harness a cornerstone of experimental design, which is.. replication! Specifically, we propose the following genomewide & data-driven framework for benchmarking algorithms 👇
28.05.2025 17:36 — 👍 0 🔁 0 💬 1 📌 0
SV detection methods have been benchmarked using small SV truth sets from cell lines. This is limited in many ways (see preprint), so the critical question now arises - how can we benchmark SV detection algorithms across the entire genome in an unbiased and data-driven manner??🤔
28.05.2025 17:36 — 👍 0 🔁 0 💬 1 📌 0
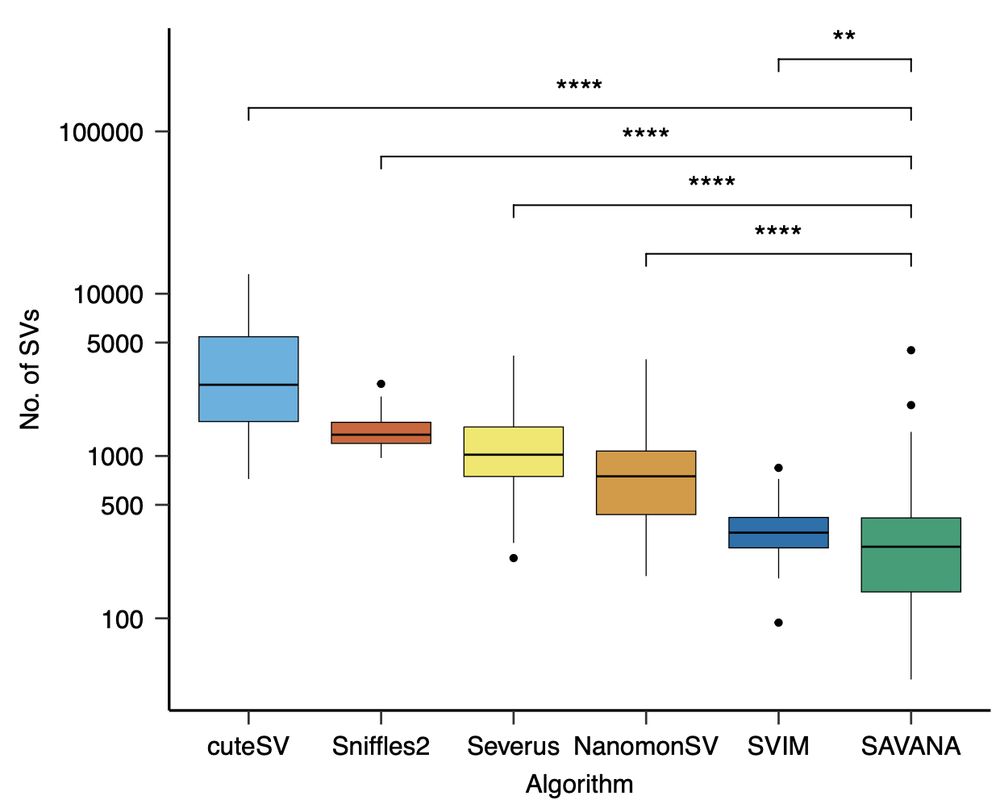
Next, we applied existing SV detection methods designed for germline SV discovery (Sniffles2, cuteSV, SVIM) and for cancer genome analysis (Severus, NanomonSV,SVision-Pro). Sv rates varied across 2 orders of magnitude – so how can we benchmark these tools?
28.05.2025 17:36 — 👍 0 🔁 0 💬 1 📌 0
Professor in computational cancer biology at UCL interested in disease, mutations, and aging. Funded by CRUK, MRC and Royal Society.
Personal account for science, code, music, photography! “Tired is the new awake”
🇨🇦 Bioinformatics Postdoc @ Francis Crick Institute & UCL | PhD @ EMBL-EBI (Cortes-Ciriano Group) 🧬 👩🏻💻 | https://github.com/helrick
Phd Student at @embl.ebi.org (Cambridge), Cortès-Cirano's group.
Previously at Université Paris-Saclay
Executive Director EMBL. I have an insatiable love of biology. Consultant to ONT and Cantata (Dovetail)
Team Leader & Independent Fellow at the @crg.eu
At the Repetitive DNA Biology (REPBIO) Lab, we leverage the latest technologies to decode nucleotide sequences for investigating how repetitive DNA shapes genome function and contributes to disease.
A scientific journal publishing cutting-edge methods, tools, analyses, resources, reviews, news and commentary, supporting life sciences research. Posts by the editors.
Research scientist at @GoogleDeepMind passionate about AI, genomics and biology.
Biology-grounded MD/PhD exec | Healthy aging & flourishing at scale | Evidence-based medicine x lifestyle × tech | Agency as engine of change | Strategic Advisor | Writer | astoundinghealthtech.com (advisory); kindwellhealth.com (agency+health)
Assistant Prof, GI Oncologist @
Duke Cancer researching immunotherapy, metastasis, and the immune microenvironment.
#FirstGen. USouthFlorida alumnus. Tufts residency survivor.
Patients don’t fail treatments, treatments fail patients.
Opinions=mine
Division of Translational Pediatric Sarcoma Research @DKFZ
Group Leader @i3S Porto; Aging and Aneuploidy Lab; Targeting #GenomicInstability in #Aging; #Rejuvenation; #Longevity; #Progeria and #Down Syndromes
Scientist. Group leader at the Sanger Institute, Cambridge UK. Somatic mutation and selection in normal tissues, cancer and ageing.
Senior Lecturer at the University of Central Lancashire. Geographer. Mum of four (two live in my heart). Tweets child loss, SUDC, PPA2 deficiency, sex work, digital-sexual exploitation, fraud, policing, VAWG. #WIASN
Associate Professor of Biomolecular Engineering at the University of California, Santa Cruz; Associate Director, UC Santa Cruz Genomics Institute
Nature Reviews Cancer is a monthly review journal for researchers working on cancer. We, the editors, highlight the most exciting topics in cancer research.
https://www.nature.com/nrc/
Professor of Biomolecular Engineering at the University of California, Santa Cruz; Associate Director, UC Santa Cruz Genomics Institute
Scientist at DKFZ and EMBL in Heidelberg, loving stats, genomics and genetics. @OliverStegle@genomic.social. For group news see @steglelab.bluesky.social
Assistant Professor @ Massachusetts General Hospital | Harvard Medical School | Broad Institute
https://vazquezgarcialab.mgh.harvard.edu/