

Opportunity for computational chemist at Inductive Bio (USA: New York City, San Francisco, or Boston) #CompChem #cheminformatics #ADMET #ChemJobs #chemsky 🧪
www.inductive.bio/careers?ashb...
Opportunity for computational chemist at Inductive Bio (USA: New York City, San Francisco, or Boston) #CompChem #cheminformatics #ADMET #ChemJobs #chemsky 🧪
www.inductive.bio/careers?ashb...
Yes, this is accurate.
21.10.2025 18:29 — 👍 11 🔁 3 💬 4 📌 0
Is this accurate?!? Re Harvard CCB
"Chemistry and Chemical Biology will go down to four or five admits, one of the professors added."
We (@sobuelow.bsky.social) developed AF-CALVADOS to integrate AlphaFold and CALVADOS to simulate flexible multidomain proteins at scale
See preprint for:
— Ensembles of >12000 full-length human proteins
— Analysis of IDRs in >1500 TFs
📜 doi.org/10.1101/2025...
💾 github.com/KULL-Centre/...

If you used our ANI MLIPs, you probably used our TorchANI library. Now, new and improved version 2.0. Use it, enjoy it, break it, let us know what you did or tried to do with it. doi.org/10.1021/acs.jcim.5c01853
@ignaciopickering.bsky.social @nickterrel.bsky.social @khuddleston.bsky.social
🔥We're excited to announce a major milestone for the machine-learned interatomic potential (MLIP) ecosystem: TorchSim is moving to community ownership and governance through a partnership with Radical AI and the open-source community! TorchSim is an atomistic simulation engine built for the AI era.
15.10.2025 19:06 — 👍 13 🔁 3 💬 1 📌 0
I received many requests to share materials from our undergraduate course “Machine Learning in Chemistry”
— here you go!
A preprint summarizing insights and lessons learned:
chemrxiv.org/engage/chemr...
A Jupyter Notebook Tutorial Gallery:
xuhuihuang.github.io/mlchem/html/...
Today my @nytimes.com colleagues and I are launching a new series called Lost Science. We interview US scientists who can no longer discover something new about our world, thanks to this year‘s cuts. Here is my first interview with a scientist who studied bees and fires. Gift link: nyti.ms/3IWXbiE
08.10.2025 23:29 — 👍 4729 🔁 1826 💬 142 📌 83
My talk at the #RDKit UGM in Prague youtu.be/TduH7v-biyY?... #compchem
05.10.2025 09:09 — 👍 15 🔁 3 💬 0 📌 0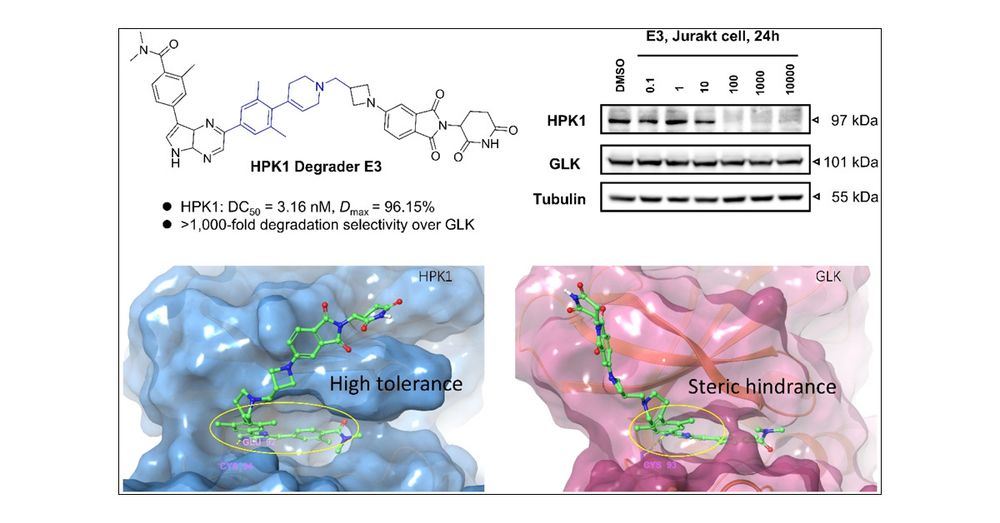
Discovery of a New and Selective HPK1 PROTAC for Enhancing Tumor Immunotherapy through Eliminating GLK Degradation
11.06.2025 11:00 — 👍 9 🔁 2 💬 0 📌 0
🧪In a JAMA viewpoint paper, the FDA expresses its intent to use AI for first-pass reviews of drug applications to significantly increase efficiency.
This is premature and we haven't seen the evidence AI is ready for this use case.
Link to the paper: jamanetwork.com/journals/jam...
#MedSky #MLSky
Someone suggested Vancouver yesterday. They were joking, but I'll bring it up next time I see Daphne
05.04.2025 17:20 — 👍 1 🔁 0 💬 1 📌 0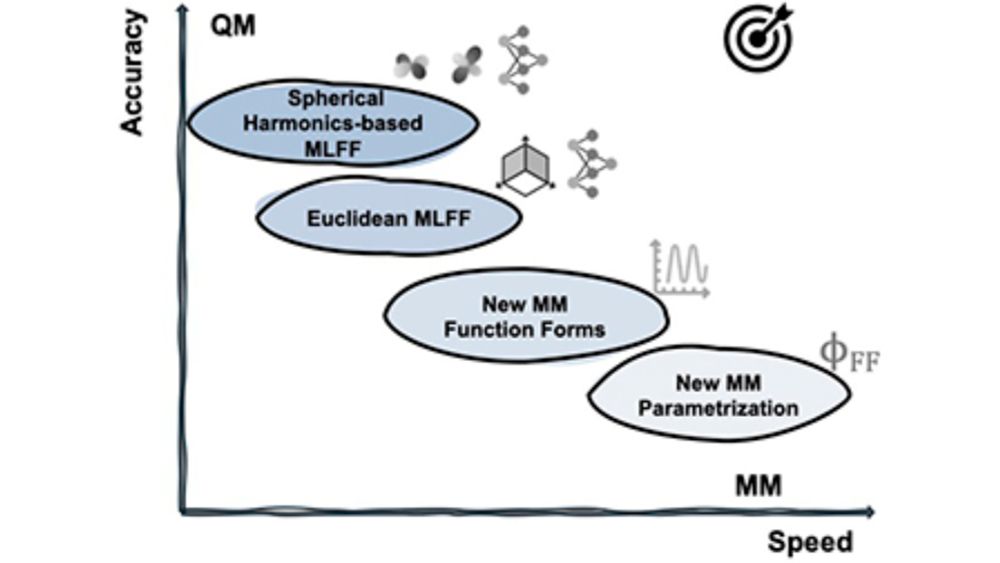
Force fields with QM accuracy and MM speed are the theoretical biophysicist's Philosopher's Stone. I used ANIX2 with OpenFF and OpenMM. The results were insightful but at the expense of huge computational costs. #MLFF #biophysics #moleculardynamics
05.04.2025 10:48 — 👍 11 🔁 6 💬 0 📌 0
Sharing slides for All-atom Diffusion Transformers
- briefly summarises the big ideas and key takeaways
Link - www.chaitjo.com/publication/...
Man, I gotta say, @aspuru.bsky.social definitely made the right choice to bail on the US way back in 2018. I mean I understood back then, but... prescient decision!!
05.04.2025 04:31 — 👍 1 🔁 0 💬 1 📌 0Incredibly grateful to have helped build the ASAP Discovery Consortium (@asapdiscovery.bsky.social) that enabled @griffen-ed.bsky.social and all the other amazing members of this team to discover a new broad-spectrum coronavirus antiviral!
21.03.2025 16:58 — 👍 23 🔁 2 💬 0 📌 0
Chemistry is changed by AI at a super rapid pace! Check our data miner for chemical reactions, MERMaid!
@accelerationc.bsky.social @thematterlab.bsky.social @shixuanleong.bsky.social #chemsky #compchemski #ai #vlm #llm #aiforscience Please follow @thematterlab.bsky.social
What an awesome group effort reporting on the structural dynamics of ~60% of available structures !!
www.nature.com/articles/s41...
not just NIH websites
02.03.2025 00:43 — 👍 7 🔁 6 💬 1 📌 0
The next step in trying to dismantle Infectious Diseases research - dismantle @niaidnews.bsky.social - should surprise no one that this a R-led proposal.
An unmitigated disaster for diagnosis, treatment and prevention of pathogens that affect everyone in the US
www.congress.gov/bill/119th-c...
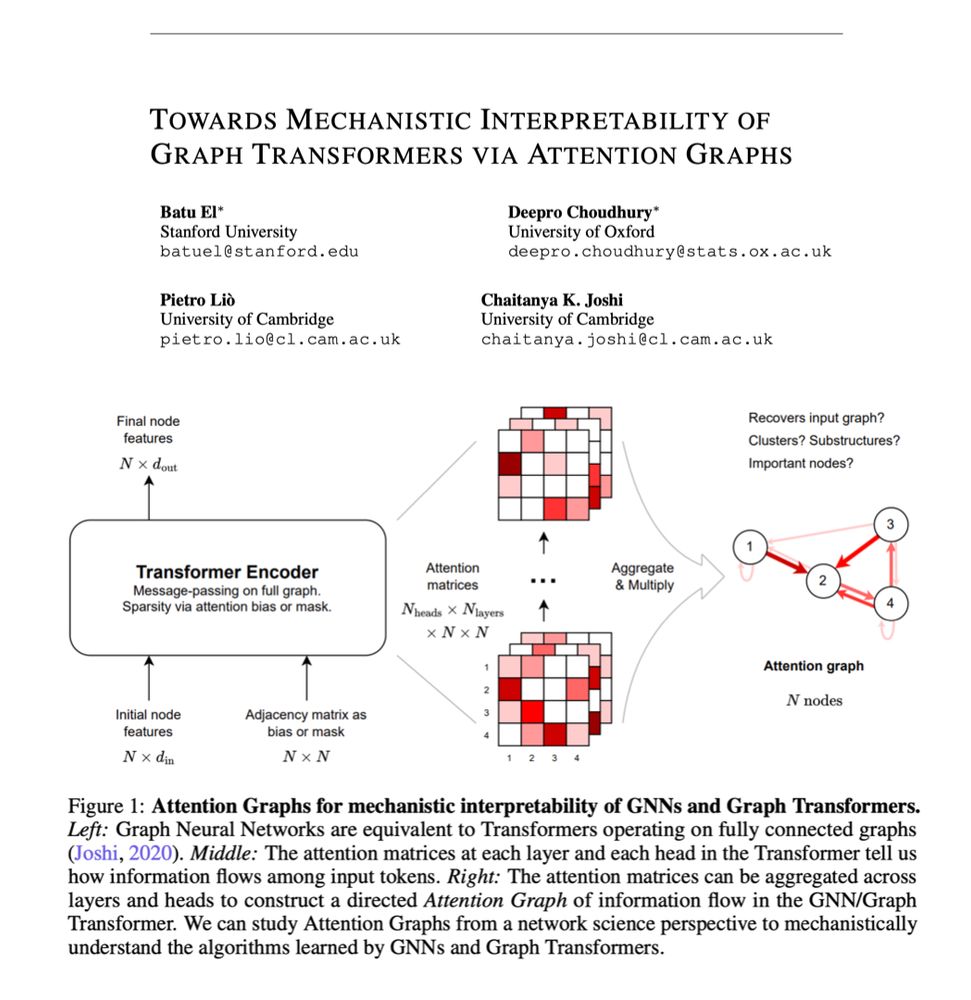
Our first attempts at mechanistic interpretability of Transformers from the perspective of network science and graph theory!
A wonderful collaboration with superstar MPhil students Batu El, Deepro Choudhury, as well as Pietro Liò as part of the Geometric Deep Learning class at @cst.cam.ac.uk
Variational Flow Matching goes Riemannian! 🔮
In this preliminary work, we derive a variational objective for probability flows 🌀 on manifolds with closed-form geodesics, and discuss some interesting results.
Dream team: Floor, Alison & Erik (their @ below) 💥
📜 arxiv.org/abs/2502.12981
🧵1/5
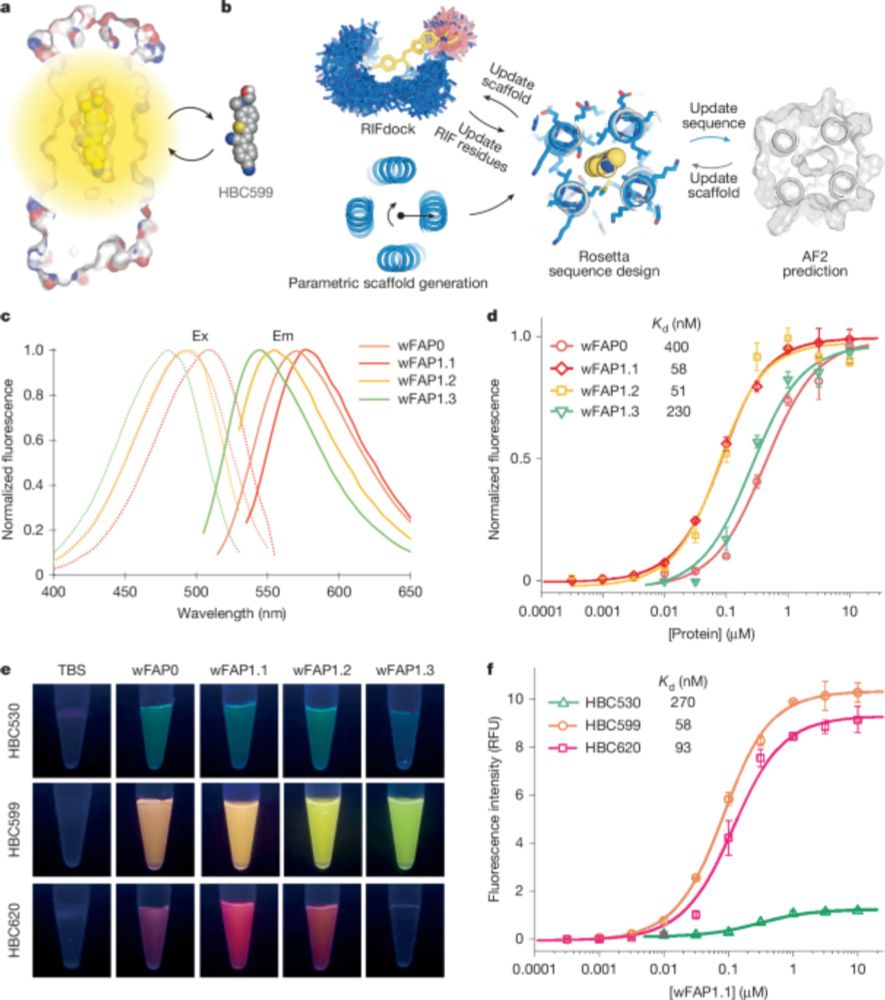
WOW!!
De novo design of transmembrane fluorescence-activating proteins
www.nature.com/articles/s41...
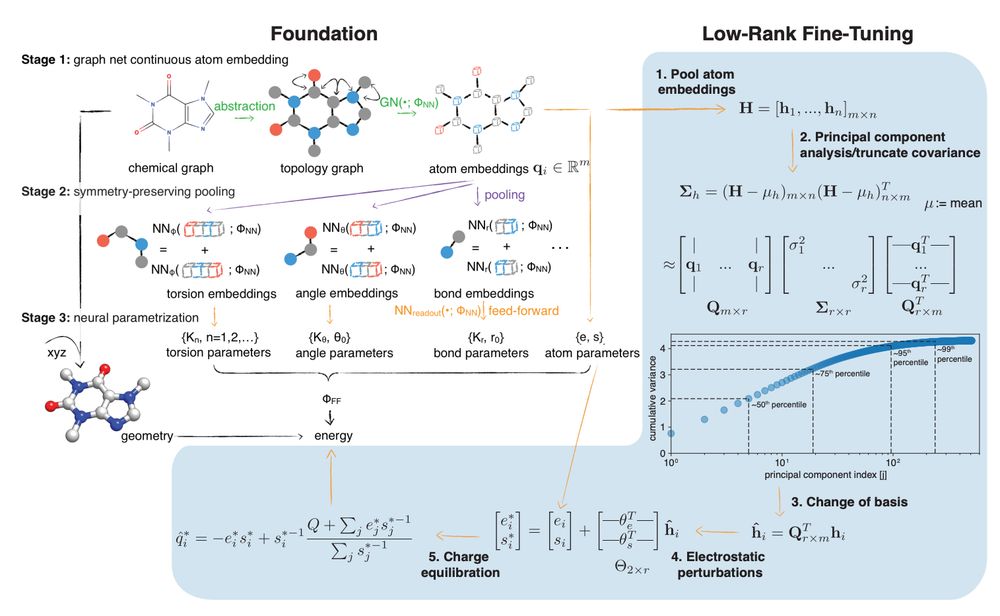
Figure 1 from arXiv preprint https://doi.org/10.1101/2025.01.06.631610 Fig. 1 Espaloma is an end-to-end differentiable molecular mechanics parameter assignment scheme for arbitrary organic molecules. Espaloma (extensible surrogate potential optimized by message-passing) is a modular approach for directly computing molecular mechanics force field parameters FFF from a chemical graph G such as a small molecule or biopolymer via a process that is fully differentiable in the model parameters FNN. In Stage 1, a graph neural network is used to generate continuous latent atom embeddings describing local chemical environments from the chemical graph. In Stage 2, these atom embeddings are transformed into feature vectors that preserve appropriate symmetries for atom, bond, angle, and proper/improper torsion inference via Janossy pooling.54 In Stage 3, molecular mechanics parameters are directly predicted from these feature vectors using feed-forward neural networks. This parameter assignment process is performed once per molecular species, allowing the potential energy to be rapidly computed using standard molecular mechanics or molecular dynamics frameworks thereafter. The collection of parameters FNN describing the espaloma model can be considered as the equivalent complete specification of a traditional molecular mechanics force field such as GAFF38,39/AM1-BCC55,56 in that it encodes the equivalent of traditional typing rules, parameter assignment tables, and even partial charge models. Reproduced from ref. 49 with permission from the Royal Society of Chemistry.
Everything is chaos, but I wanted to share some awesome recent science from the lab that hints at where the future of biomolecular simulation is headed:
Foundation simulation models that can be fine-tuned to experimental free energy data to produce systematically more accurate predictions.
The BioEmu-1 model and inference code are now public under MIT license!!!
Please go ahead, play with it and let us know if there are issues.
github.com/microsoft/bi...

Here’s the latest in the Continuing Crisis series - there will, unfortunately, be more:
18.02.2025 14:07 — 👍 42 🔁 14 💬 1 📌 1As our debut Bluesky post, we’re excited to share our new paper (first author Zachary McCaw) in HGG Advances on scrutinizing the practice of using a ratio trait (numerator / denominator) for GWAS. www.cell.com/hgg-advances...
11.02.2025 14:41 — 👍 5 🔁 2 💬 0 📌 0Very cool work!!
08.02.2025 13:16 — 👍 22 🔁 5 💬 1 📌 0