
9/n 📖 For more details, check out our updated manuscript: biorxiv.org/content/10.1...
#ProteinLM #AI4Science #ProteinDesign #MachineLearning #Bioinformatics

@chaohou.bsky.social
Protein dynamic, Multi conformation, Language model, Computational biology | Postdoc @Columbia | PhD 2023 & Bachelor 2020 @PKU1898 http://chaohou.netlify.app
9/n 📖 For more details, check out our updated manuscript: biorxiv.org/content/10.1...
#ProteinLM #AI4Science #ProteinDesign #MachineLearning #Bioinformatics

8/n 🔬 Interesting result: proteins with high likelihoods from MSA-based estimates tend to have more deleterious mutations — a pattern not observed for ESM2.
25.08.2025 02:28 — 👍 0 🔁 0 💬 1 📌 0
7/n 🔬 Interesting result: random seeds also impact predicted sequence likelihoods.
25.08.2025 02:28 — 👍 0 🔁 0 💬 1 📌 0
6/n 🔬 Interesting result: when scale up model size, predicted sequence likelihoods vary across proteins — some show no improvement, while others show substantial gains.⚡
25.08.2025 02:28 — 👍 0 🔁 0 💬 1 📌 0
5/n 🎯 pLMs align best with MSA-based per-residue likelihoods at moderate levels of sequence likelihood, which explains the bell-shaped relationship we observed in fitness prediction performance.
25.08.2025 02:27 — 👍 0 🔁 0 💬 1 📌 0
4/n 🔍 Interestingly, while overall sequence likelihoods differ, per-residue predicted likelihoods are correlated.
The better a pLM’s per-residue likelihoods align with MSA-based estimates, the better its performance on fitness prediction. 📈

3/n 🧬 Predicted likelihoods from MSA-based methods directly reflect evolutionary constraints.
Even though pLMs are also trained to learn evolutionary information, their predicted whole sequence likelihoods show no correlation with MSA-based methods. ⚡

2/n 📊 We found that general pLMs trained on large datasets show a bell-shaped relationship between fitness prediction performance and wild-type sequence likelihood.
Interestingly, MSA-based models do not show this trend.

1/n 🧬 These models estimate sequence likelihood from different inputs — sequence, structure, and homologs.
To infer mutation effects, the log-likelihood ratio (LLR) between the mutated and wild-type sequences is used. ⚖️

We just updated our manuscript "Understanding Language Model Scaling on Protein Fitness Prediction". Where we explained why larger pLMs don’t always perform better on mutation effect prediction. We extended beyond ESM2 to models like ESMC, ESM3, SaProt, and ESM-IF1.
#ProteinLM
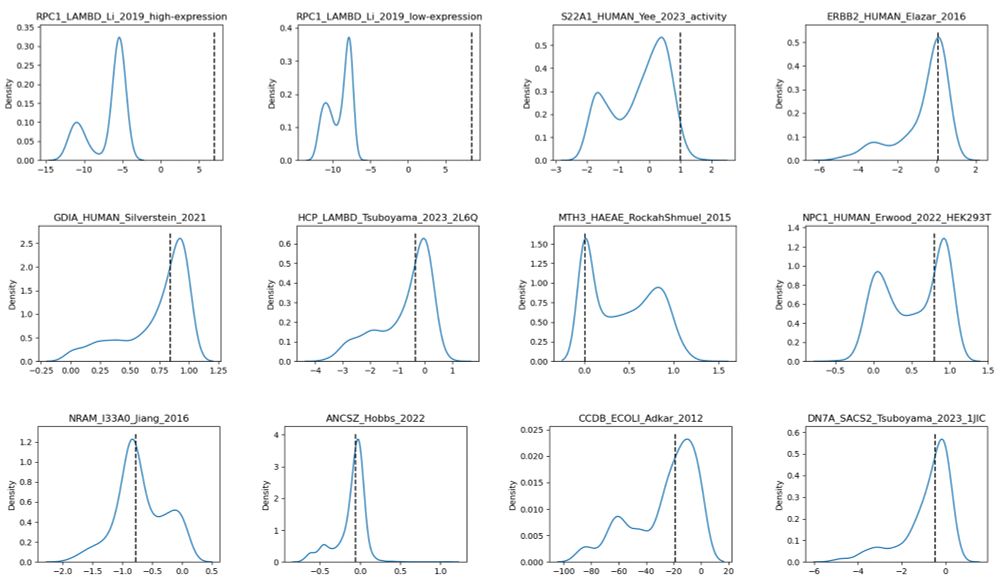
⚠️ Caution when using ProteinGYM binary classification: some DMS_binarization_cutoff values appear inverted, some are totally unreasonable given the DMS_score shows a clear bimodal distribution.
11.06.2025 15:57 — 👍 0 🔁 0 💬 0 📌 0
How can we better understand pathogenic variants in intrinsically disordered regions (IDRs)? How do models such as AlphaMissense and ESM1b predict pathogenicity, when these regions typically exhibit lower genomic conservation than ordered regions? Read more:
doi.org/10.1101/2025...
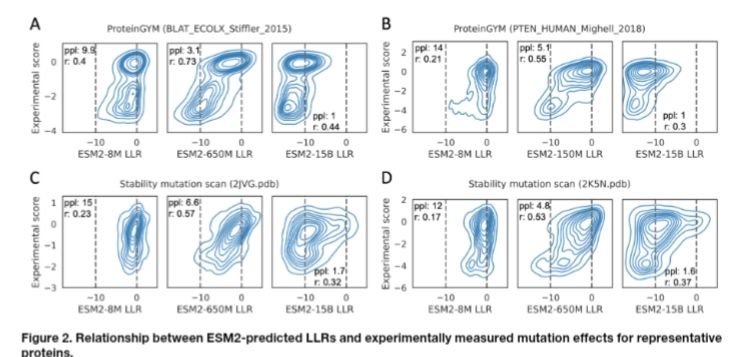
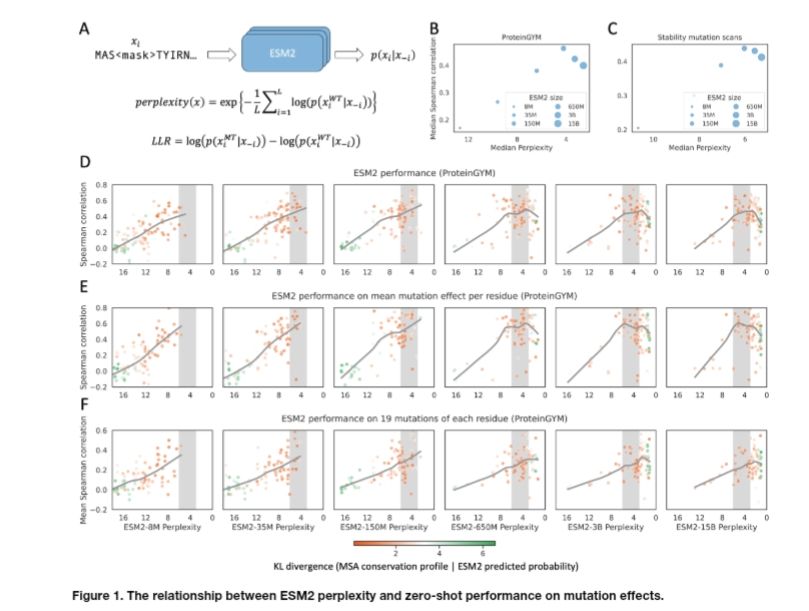
Relationship between perplexity and zero shot performance.
Protein language model likelihood are better zero shot mutation effect predictions when they have perplexity 3-6 on the wildtype sequence.
www.biorxiv.org/content/10.1...

read our preprint here: www.biorxiv.org/content/10.1...
29.04.2025 17:55 — 👍 0 🔁 0 💬 0 📌 0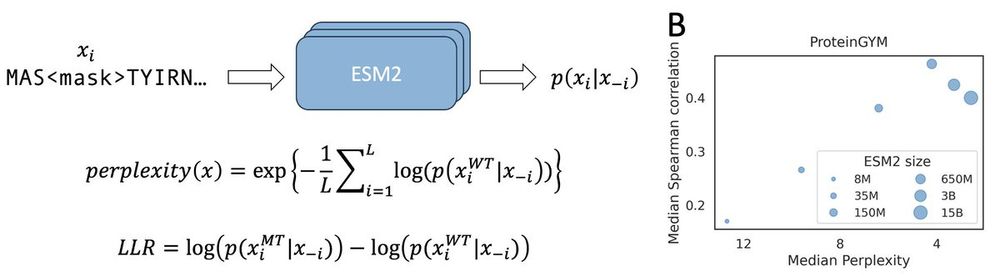
Why do large protein language models like ESM2-15B underperform compared to medium-sized ones like ESM2-650M in predicting mutation effects? 🤔
We dive into this issue in our new preprint—bringing insights into model scaling on mutation effect prediction. 🧬📉
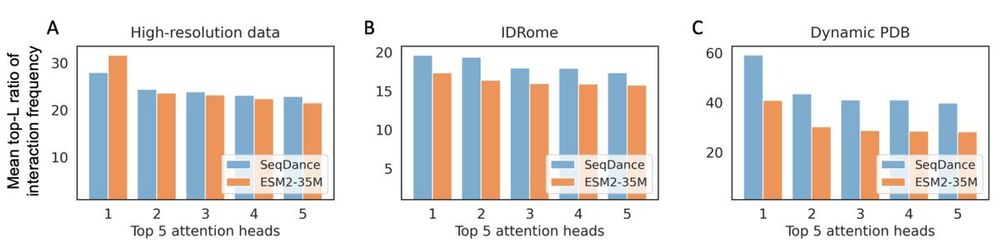
4/n We also compared SeqDance's attention with ESM2-35M.
17.04.2025 14:43 — 👍 0 🔁 0 💬 0 📌 0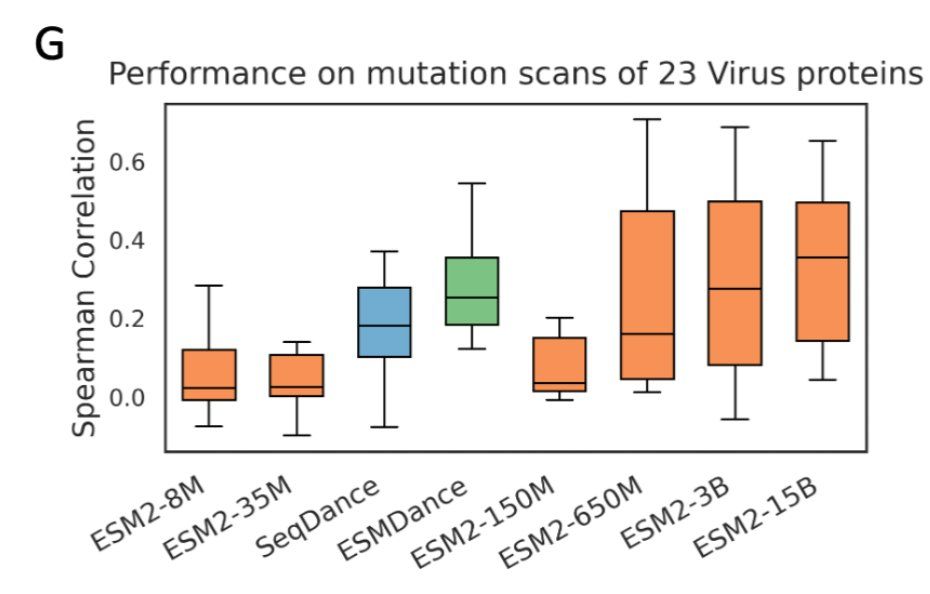
3/n here is the comparison on viral proteins in ProteinGYM, (our models have 35M parameters)
17.04.2025 14:43 — 👍 0 🔁 0 💬 1 📌 0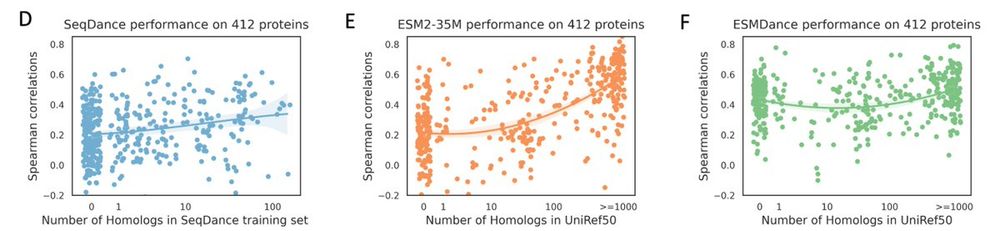
2/n on the mega-scale protein stability dataset, it's clear that ESM2's performance is correlated with the number of homologs in its training set. but our models show robust performance for proteins without homologs in training set.
17.04.2025 14:42 — 👍 0 🔁 0 💬 1 📌 0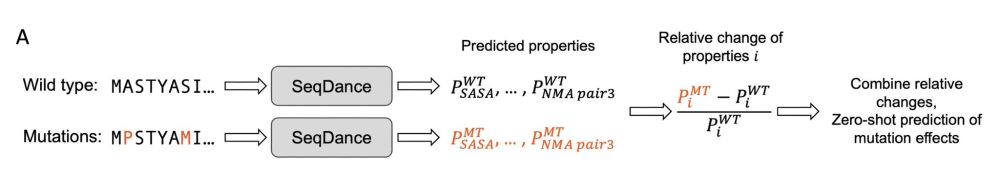
1/n to perform zero-shot fitness prediction, we use our models SeqDance/ESMDance to predict dynamic properties of both wild-type and mutated sequences. the relative changes bettween them are used to infer mutation effects.
17.04.2025 14:41 — 👍 0 🔁 0 💬 1 📌 0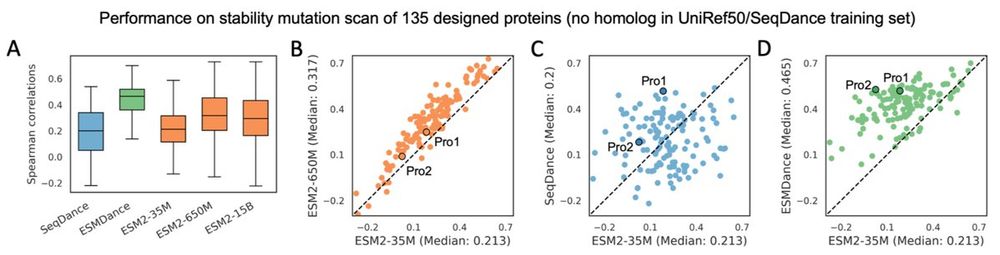
We have updated our protein lanuage model trained on structure dynamics. Our new models show significant better zero-shot performance on mutation effects of designed and viral proteins compared to ESM2. check the new preprint here: www.biorxiv.org/content/10.1...
17.04.2025 14:40 — 👍 2 🔁 2 💬 1 📌 0
SeqDance: A Protein Language Model for Representing Protein Dynamic Properties https://www.biorxiv.org/content/10.1101/2024.10.11.617911v1
15.10.2024 16:49 — 👍 1 🔁 1 💬 0 📌 0


















