

For our own bingo card with @enrimassi.bsky.social, scrappy crappy trophy. Only a single rare!
07.02.2026 17:52 — 👍 2 🔁 0 💬 0 📌 0
For our own bingo card with @enrimassi.bsky.social, scrappy crappy trophy. Only a single rare!
07.02.2026 17:52 — 👍 2 🔁 0 💬 0 📌 0
Our latest paper introduces new methods for improving peptide retention time predictions in proteomics, incorporating chemical structure information to better handle unseen modifications.
Read all about it in our preprint by👉 doi.org/10.1101/2025...
#Proteomics #AI #MachineLearning

Exciting news: Preprint on the limitations of current de novo peptide sequencing models on dealing with sequence ambiguity is now out! It focuses on how current models deal with sequence ambiguity, and when and where they go wrong.
Check it out here: www.biorxiv.org/content/10.1...
Thanks (especially as I was so vague). It feels like a lot of scripting for sure. After chatting with Magnus, even working through some tutorials like those collabs by @robbinbouwmeester.bsky.social on ProteomicsML wouldn't be a bad idea.
25.06.2025 16:46 — 👍 4 🔁 1 💬 1 📌 0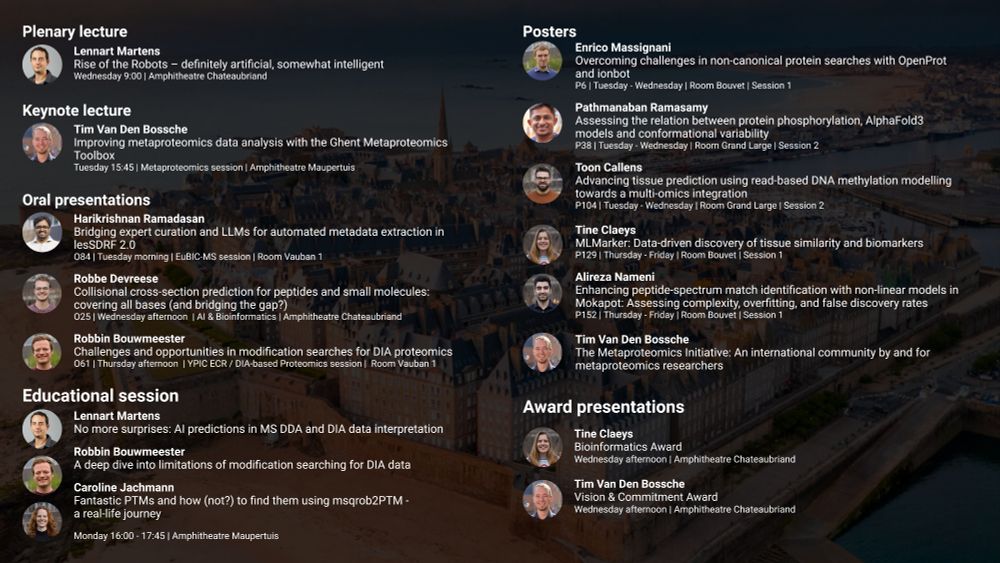
Plenary talk Lennart Martens Rise of the Robots – definitely artificial, somewhat intelligent Keynote lecture Tim Van Den Bossche Improving metaproteomics data analysis with the Ghent Metaproteomics Toolbox Oral presentations Harikrishnan Ramadasan Bridging expert curation and LLMs for automated metadata extraction in lesSDRF 2.0 Robbe Devreese Collisional cross-section prediction for peptides and small molecules: covering all bases (and bridging the gap?) Robbin Bouwmeester Challenges and opportunities in modification searches for DIA proteomics Educational session Lennart Martens No more surprises: AI predictions in MS DDA and DIA data interpretation Robbin Bouwmeester A deep dive into limitations of modification searching for DIA data Caroline Jachmann Fantastic PTMs and how (not?) to find them using msqrob2PTM - a real-life journey Poster presentations Enrico Massignani Overcoming challenges in non-canonical protein searches with OpenProt and ionbot Pathmanaban Ramasamy Assessing the relation between protein phosphorylation, AlphaFold3 models and conformational variability Toon Callens Advancing tissue prediction using read-based DNA methylation modelling towards a multi-omics integration Tine Claeys MLMarker: Data-driven discovery of tissue similarity and biomarkers Alireza Nameni Enhancing peptide-spectrum match identification with non-linear models in Mokapot: Assessing complexity, overfitting, and false discovery rates Tim Van Den Bossche The Metaproteomics Initiative: An international community by and for metaproteomics researchers Award presentations Tine Claeys Bioinformatics Award Tim Van Den Bossche Vision & Commitment Award
From PTMs to proteins, from metadata to metaproteomics. CompOmics has got you covered at #EuPA2025!
16.06.2025 08:05 — 👍 22 🔁 8 💬 0 📌 0
And you get the cutest mascotte octopus. His name is Mark! He'll guide you Clippy-wise through your analyses 🙌
16.06.2025 12:32 — 👍 8 🔁 1 💬 2 📌 0
MLMarker is live! This ML-tool predicts tissue similarity and uncovers biomarkers from your proteomics data. It was trained on public data of healthy human tissues.
Preprint & app: www.biorxiv.org/content/10.1...
Let's chat at #EuPA2025 - Award session (Wednesday) & poster session (Thursday)!
We recently released a tool to help you with this. 🚀 Say hello to pridepy — your Python for grabbing data from the @pride-ebi.bsky.social!
To search metadata or download files via FTP, Aspera, Globus, or S3, and is perfect for bioinfo workflows.
Check it out 👉 github.com/PRIDE-Archiv...

New in DeepLC! Ability to deal with wild, weird, and wobbly LC setups or peptide modifications. This ability is possible with transfer learning; where only a minimal amount of training peptides are needed for accurate retention time predictions.
www.biorxiv.org/content/10.1...
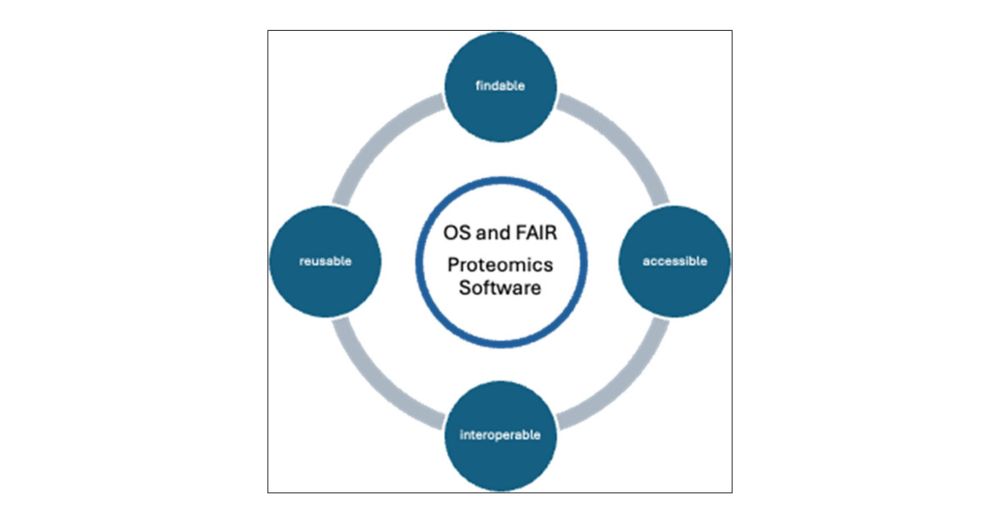
Fantastic review with an unusual history, growing out of a passionate blog post by @willfondrie.com (willfondrie.com/2024/10/the-...), resulting from a storm (in our teacup) on X during @hupo-org.bsky.social 2024. Great teamwork, authors! pubs.acs.org/doi/10.1021/...
24.04.2025 18:24 — 👍 16 🔁 11 💬 0 📌 1Looks amazing 😍
24.03.2025 11:51 — 👍 1 🔁 0 💬 1 📌 0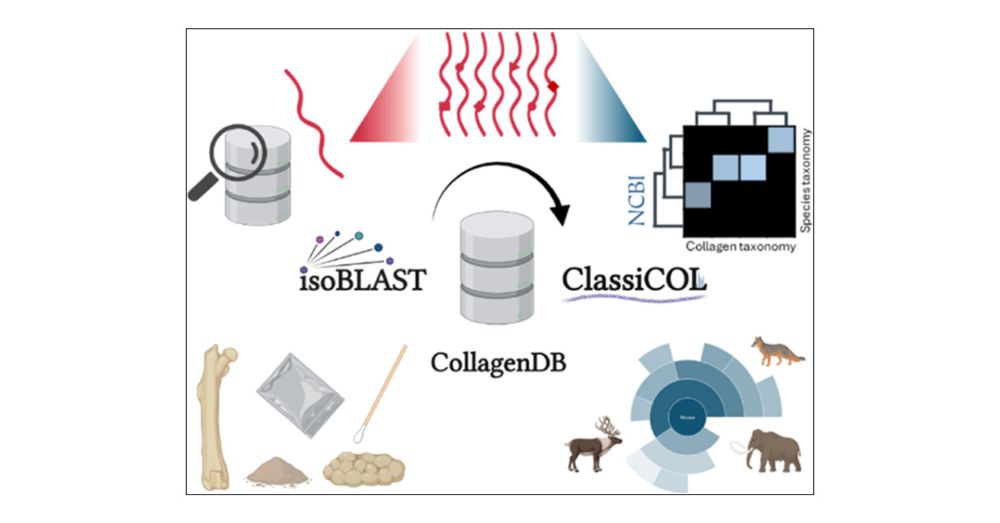
Classification of Collagens via Peptide Ambiguation, in a Paleoproteomic LC-MS/MS-Based Taxonomic Pipeline #JProteomeRes pubs.acs.org/doi/10.1021/...
14.03.2025 08:17 — 👍 4 🔁 3 💬 0 📌 0
MS2Rescore found its way into #ProteomeDiscoverer 🥳 At least, somewhat, through #MascotServer. Thanks for the implementation and for the nice blog post, @matrixscience.bsky.social!
www.matrixscience.com/blog/using-m...
What excites me most is that it introduces the first ML-based solution for peptide multiconformers. But that’s not all! We also demonstrate a substantial performance boost for uniconforming peptides.
Our findings are clear: multiconformer peptides cannot be overlooked when predicting CCS!

🚀 New preprint alert! We've improved IM2Deep for accurate peptide collisional cross-section (CCS) prediction, even for peptides exhibiting multiple conformations in the gas phase! 🎯
Check it out here:
www.biorxiv.org/content/10.1...

DIA-NN 2.0 is released! We consider it the biggest step forward in the history of DIA-NN. On modern LC-MS almost all identifications are now peptidoform-confident, with major improvements e.g. for phospho. Some other cool things too: github.com/vdemichev/Di...
29.01.2025 09:04 — 👍 150 🔁 39 💬 6 📌 2Or even better, use the transfer learning ability of DeepLC with a good base model (e.g., the one above)
19.12.2024 14:26 — 👍 0 🔁 0 💬 0 📌 0Cool! Small comment, indeed the hela_hf model can predict from TMT-labelled peptides, it needs to extrapolate a lot. Best is probably to use this model: github.com/RobbinBouwme...
19.12.2024 14:25 — 👍 1 🔁 0 💬 1 📌 0
The end of the year always comes with many 3D print requests. Hope the soon to be PhD will enjoy this ornament :)
16.12.2024 08:53 — 👍 15 🔁 0 💬 0 📌 0
Recently, We saw a discussion on the role of open-source in proteomics. Here, experienced developers & researchers maintaining OS tools for years shared this comment to guide newcomers in the field about OS and its role in the field. 💻 #Proteomics #OpenSource chemrxiv.org/engage/chemr...
09.12.2024 13:03 — 👍 42 🔁 23 💬 1 📌 4
This was a ton of fun to write with @ypriverol.bsky.social and all of the other authors 👏
Our goal was to share a vision of #OSS #proteomics for us to build toward, and propose some ways to get there 🚀
I’m blown away by how many folks contributed and how much it evolved beyond just my voice 🙌

Caroline Jachmann presenting her work in front of the BePAc 2024 audience

Robbe Devreese presenting his work at BePAc 2024
Today, @robbedevr.bsky.social and @carojachmann.bsky.social presented their work on #IM2Deep and #ProteoBench at #BePAc2024.
Learn more at doi.org/10.1101/2024... and proteobench.readthedocs.io.
Looks 3D printed, are you sure this is food-safe 😬. There is a lot of discussion around this, especially because of the grooves and edges that easily catch food and are hard to clean. Be sure to clean it very well, and maybe run some swabs of the cutter on your timsTOF ;)
26.11.2024 10:03 — 👍 3 🔁 0 💬 1 📌 0Although I might be biased, in my opinion it is the best stream-lined experience for rescoring. Even going to quant with tools such as FlashLFQ, simply works phenomenally.
30.01.2024 09:35 — 👍 1 🔁 0 💬 0 📌 0I think it was Robbin Bouwmeester’s talk at EuBIC where it was SAGE to MS2Rescore and it seemed quietly bad ass. This update is making it look even more attractive as my new fav proteomics pipeline.
29.01.2024 12:52 — 👍 5 🔁 4 💬 1 📌 0