Naive question: I thought targeted was about sensitivity -
why ng?
03.01.2026 11:33 — 👍 2 🔁 0 💬 1 📌 0
It's called the Agilent 6495D.
03.01.2026 10:32 — 👍 5 🔁 1 💬 1 📌 0
Not a conference, but worth flagging:
EMBO Practical Course “Targeted proteomics: advanced tools for biomedical research” Barcelona, 8–13 Nov 2026
Line-up not announced yet, but previous editions organized by @maccoss.bsky.social with invited speakers incl. Alexey Nesvizhskii and Vadim Demichev.
27.12.2025 23:38 — 👍 5 🔁 1 💬 1 📌 0
Proteomic Ruler question:
In Wiśniewski et al., MCP 2014, the histone→DNA proxy seems implicit.
Is there any explicit reference stating that the Ruler uses only core histones (H2A/H2B/H3/H4) and excludes H1?
#proteomics #massspec
14.12.2025 13:41 — 👍 2 🔁 2 💬 0 📌 0

I agree MaxLFQ isn’t meant for absolute quantification.
But that still doesn’t explain the complete lack of correlation with UPS2.
From my experience, iBAQ and MaxLFQ usually correlate well (R² ~0.78, non related example dataset shown), suggesting they track the same MS1 signal.
07.12.2025 23:00 — 👍 1 🔁 0 💬 1 📌 0
As shown in the DIA-NN paper, the mobility term contributes only negligibly to the discriminant score, suggesting that measured CCS -even with good IM resolution - might simply be too affected by gas-phase ion–ion / ion–neutral interactions to provide a stable, high-specificity constraint …
06.12.2025 14:16 — 👍 4 🔁 0 💬 0 📌 0
With a quadrupole we know exactly which precursor m/z window was isolated -the precursor mass is tightly defined with a well-characterised error. My question was whether CCS can provide anything close to that level of search-space restriction for database searching.
06.12.2025 14:12 — 👍 2 🔁 0 💬 1 📌 0
My point rather was whether precursor CCS can actually constrain the search space during database searching not just what Da-equivalent tolerance it has.
06.12.2025 14:04 — 👍 1 🔁 0 💬 1 📌 0
Asking for a friend: Is intrinsic specificity of CCS high enough to serve as an effective in silico precursor filter during database searching?
06.12.2025 12:24 — 👍 3 🔁 0 💬 1 📌 0
“Very interesting! Do you know whether ProteomeSciences is already testing the new DXT tags with selected customers or collaborators, or is it still entirely in-house at this stage?”
20.11.2025 11:54 — 👍 1 🔁 0 💬 1 📌 0
Bonus, info about DIA multiplex tags, up to 30-plex:
"trademark DXT for our DIA multiplex tags...advances have been made in DXT multiplexing since ASMS with the number of tags increased from 6 to 11 and with the potential to
increase these to beyond 30"
18.11.2025 10:09 — 👍 2 🔁 1 💬 1 📌 0
Which LC & Flow?
07.10.2025 16:38 — 👍 1 🔁 0 💬 0 📌 0

Surprised that u go so low. With EvoSep 24 min method we can load lots more on our Ultra2 until reaching saturation especially with ICC2.0
07.10.2025 14:56 — 👍 0 🔁 0 💬 1 📌 0
How much are you loading per injection? Is ICC 2.0 enabled on the Ultra2? And which library are you using?
07.10.2025 06:01 — 👍 1 🔁 0 💬 1 📌 0
True — but the odd part is that the Human Reference Proteome is not really ‘canonical only’. Non-canonical entries from TrEMBL are included, yet the curated SwissProt isoforms are missing default. That’s what undermines the idea of a high-quality reference set.
02.10.2025 06:34 — 👍 0 🔁 0 💬 1 📌 0

On a separate note: I was surprised to find that none of the non-canonical SwissProt isoforms are included in the official human reference proteome (UP000005640).
Anyone know what’s going on here? 🤔
#proteomics #bioinformatics @pwilmarth.bsky.social il
01.10.2025 15:13 — 👍 2 🔁 0 💬 2 📌 0

Hey #TeamMassSpec,
Many non-human proteomics studies still search against taxon-filtered FASTAs.
❌ Redundant sequences
❌ Inflated search space
✅ Reference proteomes cut redundancy, improve annotation, and make results comparable.
👉 Time to move beyond taxon filters. #proteomics #massspec #uniprot
30.09.2025 14:21 — 👍 9 🔁 2 💬 2 📌 0

Without #2, a lower ion count is needed just to be sure that the full MS range is scanned, but with more accurate ion counts, you can go to the max S/N without losing ions on the edges.
This could also work for the Orbitrap Astral.
Bonus: DIAPASEF on Thermo - patentscope.wipo.int/search/en/de...
28.09.2025 13:25 — 👍 8 🔁 2 💬 2 📌 0

With 𝗗𝗜𝗔-𝗡𝗡 𝟮.𝟯.𝟬 Preview (Academia-only for now), we showcase the transformative new capabilities that have been developed in the past months. Download: github.com/vdemichev/Di...
26.09.2025 09:52 — 👍 29 🔁 6 💬 3 📌 0

Thanks @pwilmarth.bsky.social - also included the less-redundant "one protein per gene" db here ...Has anybody assessed potential benefits of the reduced search space on sensitivity?
25.09.2025 21:21 — 👍 2 🔁 0 💬 2 📌 0

aaah guess its "hidden" there :)
25.09.2025 13:39 — 👍 2 🔁 0 💬 1 📌 0
Great thanks! Where can I find the one protein per gene option?
25.09.2025 13:34 — 👍 0 🔁 0 💬 2 📌 0

Hey #TeamMassSpec,
When you run proteomics on non-human species (mouse, rat, macaque, etc.) — which protein FASTA do you prefer?
Taxonomy-filtered UniProt (all entries)
Reference proteome (SwissProt+TrEMBL)
Ensembl/GENCODE
Something else?
25.09.2025 12:41 — 👍 4 🔁 0 💬 1 📌 0
💯
06.08.2025 21:07 — 👍 1 🔁 0 💬 0 📌 0
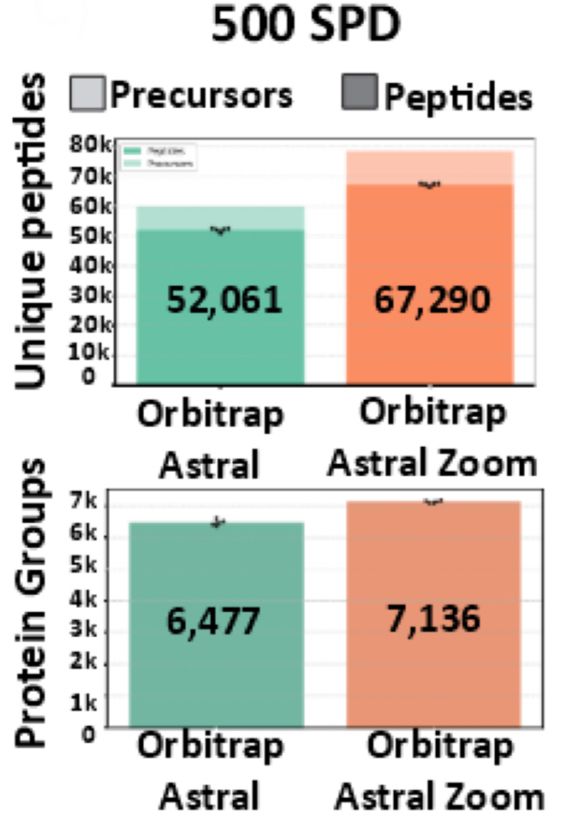
Astral Zoom hits >7,000 protein groups & 67,000 precursors — on a 500 SPD EvoSep ENO run.
www.biorxiv.org/content/10.1...
19.07.2025 09:01 — 👍 5 🔁 0 💬 0 📌 0
Is that narrow windowed thin-PASEF?
09.07.2025 21:49 — 👍 0 🔁 0 💬 1 📌 0
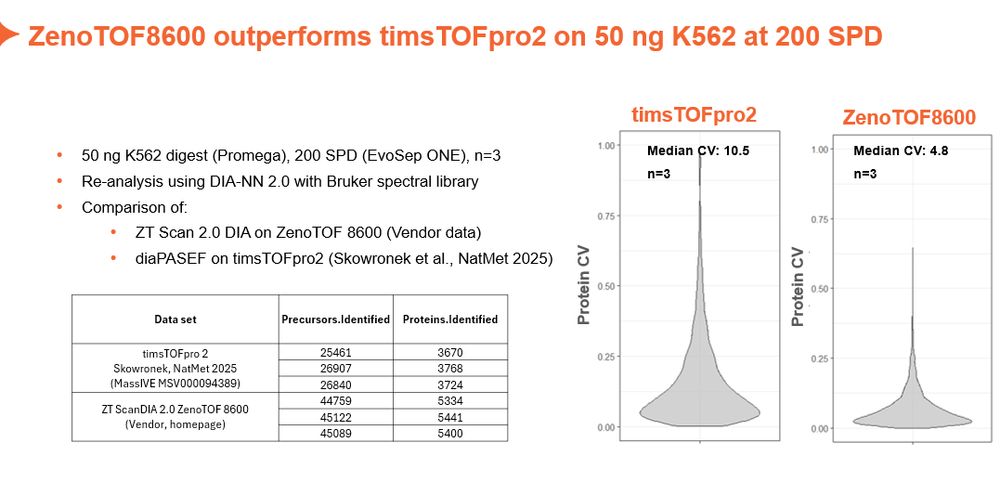
Sciex in the game.
Pretty impressed by ScanningSWATH data on the new ZenoTOF 8600.
#TeamMassSpec
27.06.2025 10:13 — 👍 11 🔁 2 💬 1 📌 0
The home of terrible maps with a pinch of humour
terriblemaps@gmail.com
Old person forgetting everything they ever knew about proteomics at speeds in excess of 270 Hz...
PostDoc @RalserLab #systemsbiology, #lcms, structural and quantitative high-throughput #proteomics+PTMs. Past @DemichevLab & @RappsilberLab
i learned more from a three minute record than i ever learned from a large language model.
Meme Magic / SocMed Psy / AI / Climate / Ex-Nerdcore.de
http://goodinternet.substack.com
http://goodmusic.substack.com
https://sigmoid.social/@rawx
A nonprofit research institute for advancing protein research in human health and disease | parallelsq.org
Computational biologist & blogger
Scientist and software developer. Creator of tidyplots.org. Website jbengler.de.
#Bioinformatics PhD Student currently focusing on #immunopeptidomics pipeline development @nf-co.re 🧬👨💻
Mass spectrometrist and facility manager interested in all things non targeted.
Loves unifying art and science with philosophy. I study proteins on a spatiotemporal canvas.
https://sites.google.com/view/bharathabnair/home
Electric biomancer: Microbiologist turned mass spectrometrist with a hefty emphasis on all things proteomics!
Bioinformatician • UTDallas, previously UniOxford | pain, proteomics, multi-omics & open science | chronic migraines | raised on unceded Algonquin, Anishinabek territory | she/they 🌈🍉
Group leader at Leibniz-Forschungsinstitut für Molekulare Pharmakologie
W2-S Professor at Charité – Universitätsmedizin Berlin
PhD scientist training in proteomics and AAV gene therapy in the Lilley lab, University of Cambridge 🧪🧬❤️ Also a big fan of scuba diving, cooking and espresso martinis. (She/Her).
Archaeology, University of Cambridge | The Globe, University of Copenhagen
Ancient Proteins | Medieval Manuscripts | Proteomics and AI | 🇺🇦
Senior Lecturer in Palaeoproteomics at the University of York
PhD at Utrecht University
Proteomics | Immuno-oncology
Genomics & Proteomics & Democracy enthusiast. Extracellular Vesicles, Oncology, Early Detection, Screening. Opinions are my own. 🇩🇪in MA, 🇺🇸
⚽️⚪️🔴🐊 ⚓️
ORCID:0009-0005-7342-3279