"Probing the Dark Energy in the Functional Protein Universe" is now published in @pnas.org 🎉
www.pnas.org/doi/10.1073/...
22.01.2026 19:09 — 👍 6 🔁 4 💬 0 📌 0


Our method for predicting folding dynamics/mechanisms from sequence info is now a book chapter! link.springer.com/book/9781071...
@diegulise.bsky.social
06.11.2025 12:12 — 👍 1 🔁 1 💬 0 📌 0
Applications are open for the @crg_eu PhD Programme! 20 fully funded positions — including one in our group through the Evolutionary Medical Genomics ITN.
Join us to develop deep generative models of cross-species data to tackle open questions in disease genetics.
www.crg.eu/en/content/t...
23.10.2025 11:00 — 👍 11 🔁 9 💬 0 📌 3

Google Colab
Dark Energy is not a relative score, but provides a common scale and has physical energy units. And it can be also computed even if ΔΔG is not available, using AWSEM force-field. You can compute it for your favorite PDB colab.research.google.com/github/eagalpern/colabs/blob/main/DarkEnergy.ipynb
12.08.2025 14:49 — 👍 1 🔁 0 💬 0 📌 0

Some sites have, on average over all the possible variants, high Dark Energy. In those cases, there is a function beyond protein folding affecting natural selection. This is clearly the case of Enzyme catalytic sites
12.08.2025 14:49 — 👍 1 🔁 0 💬 1 📌 0

The idea is simple. For a deleterious mutation, we expect the protein to get destabilized. But how much? Correlations between a ΔΔG DMS and the corresponding ESM-2 scores are not perfect. We define the difference between these two free energies as a ‘Dark Energy’.
12.08.2025 14:49 — 👍 0 🔁 0 💬 1 📌 0

Probing the Dark Energy in the Functional Protein Universe
We show how to localize and quantify the functional evolutionary constraints on natural proteins. The method compares the perturbations caused by local sequence variants to the energetics of the prote...
❗New Preprint w/ C Bueno (@cabb99.bsky.social), IE Sánchez (@nachoquique.bsky.social), PG Wolynes & DU Ferreiro (@diegulise.bsky.social)
We define the Protein Dark Energy and use it to localize and quantify the functional evolutionary constraints on natural proteins
arxiv.org/abs/2508.08109
12.08.2025 14:49 — 👍 12 🔁 5 💬 1 📌 2
Google Colab
And you can make folding mechanism predictions for your favorite protein!
colab.research.google.com/github/eagal...
15.07.2025 20:57 — 👍 0 🔁 0 💬 0 📌 0

Also, we show that both the stability and cooperativity changes induced by mutations can be computed directly using sequence-based evolutionary models.
6/n
15.07.2025 20:57 — 👍 2 🔁 0 💬 2 📌 0
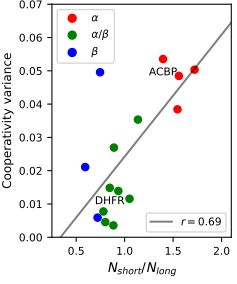
Protein topology imposes limits on the variability of folding cooperativity within a family. While most beta and alpha/beta structures exhibit only a few possible mechanisms despite high sequence diversity, alpha topologies allow for diverse folding scenarios.
5/n
15.07.2025 20:57 — 👍 0 🔁 0 💬 1 📌 0

For 15 diverse protein families, we computed the folding mechanisms of hundreds of proteins by simulating an Ising chain of folding elements, or foldons. The energetics is determined by each amino acid sequence.
4/n
15.07.2025 20:57 — 👍 0 🔁 0 💬 1 📌 0

We show that it is possible to use sequence info to go beyond predicting native structures and global stability to infer the folding mechanisms of globular proteins. We mapped a Potts evolutionary energy at the amino-acid level to a coarse-grained description of folding.
3/n
15.07.2025 20:57 — 👍 0 🔁 0 💬 1 📌 0
We know that closely related proteins usually share similar three-dimensional structures. But differences in their amino acid sequences can lead to distinct folding mechanisms, enabling the natural evolution of diverse biological functions.
2/n
15.07.2025 20:57 — 👍 0 🔁 0 💬 1 📌 0
Finally published!
“Inferring protein folding mechanisms from natural sequence diversity”
Open acces link: authors.elsevier.com/a/1lQvh1SPTB...
@biophysj.bsky.social
A new chapter of an amazing project w/ @diegulise.bsky.social and Ernesto Roman
1/n
15.07.2025 20:57 — 👍 6 🔁 0 💬 1 📌 0
Biochemist/Biophysicist. Love eating, mastering games and learning.
Scientist & artist. Prof at UMass Chan Med School & Broad Institute. Rice U alum. Founder of Zoonomia & DarwinsArk.org. Wants your dog’s DNA. And your cat’s! 🇸🇪🇳🇿🇺🇸
Organizing the largest workshop concerning Free Energy calculations within the United States.
Part of Wellcome Sanger Institute and funded by Wellcome, we develop and deliver genomics-focused learning and training. Our events support scientific careers, as well as the equitable application of genomics into research and healthcare across the globe.
Interested in all things ML+Bio especially protein design
PhD in Pharmacology
https://sites.google.com/view/vibilab
CIBION - CONICET
🇦🇷 🇫🇷 🇮🇹
physicist by training, reporter by trade
writer by nature
📍chicago
tips: katrina.miller@nytimes.com
signal: katrinamiller.04
https://katrinamillerphd.com/
protein biochemistry and evolution | viruses and antibodies | faculty @uofubiochem.bsky.social
starr.biochem.utah.edu
Playing with #singlemolecules, bending light, and algorithms to see really small things. Led by Matthew Lew, Associate Professor @WashUESE @WashUengineers @WashU #superresolution #microscopy #SMLM #computationalimaging
Schmidt Science Fellow | Postdoc @ Stanford | Prev. DPhil @ Oxford || AI for Molecule & Cell Modeling
David Bartel's lab @WhiteheadInst @MIT @HHMI | microRNAs, mRNAs, and other RNAs
Protein folding nerd, still a grad student (at my age? yes!!) UW IPD. She/any and AutAF
Computational biophysicist - enhanced sampling molecular dynamics, membrane proteins, computer-aided drug discovery
Further interests in philosophy of science, science communication and jazz music
🇺🇦 Associate professor UCONN Health. NMR, structural biology, ubiquitination, sumoylation, DUBs, sciArt
Student-run THG Lab account @UC Berkeley. We develop physics-based and machine learning-based models for various systems.
#MolecularSimulations | Associate Professor Tel Aviv University | PostDoc: Rothschild Fellow, Parrinello group, ETH Zurich | PhD: Adams Fellow, w/Benny Gerber, Hebrew University
Asst. Prof of Chemistry🧪 & Biophysics 🧬 at JohnsHopkins; unofficial lab rabbi ✡️; ally 🏳️🌈; #TeamMassSpec. PI of structural proteomics/protein folding/ageing research lab.
grigoriefflab.umassmed.edu/