
The analogy I find myself frequently using here: if everyone smoked (or no one did), the heritability of lung cancer would go up. We think about this a lot in the context of cross-biobank comparisons
31.01.2026 01:07 — 👍 6 🔁 0 💬 0 📌 0The analogy I find myself frequently using here: if everyone smoked (or no one did), the heritability of lung cancer would go up. We think about this a lot in the context of cross-biobank comparisons
31.01.2026 01:07 — 👍 6 🔁 0 💬 0 📌 0gs://ukb-diverse-pops-public/misc/pairwise/pairwise_correlations_regressed.txt.bgz - it’s coded in the way that our pan-UKB phenotypes were so not sure if it’s super easy to use but that’s pairwise r_p for ~14k phenos
08.10.2025 18:01 — 👍 0 🔁 0 💬 0 📌 0Special thanks to all co-authors that got this here including @masakanai.bsky.social @rahulg603.bsky.social @dalygene.bsky.social @egatkinson.bsky.social and of course @genetisaur.bsky.social for driving this through 5 years of work (after the first GWASes were done!)
18.09.2025 17:32 — 👍 4 🔁 0 💬 0 📌 0Tons of lessons learned around carefully controlling population stratification, using heritability as a QC metric, and probably most importantly, quantifying novelty in a mega-phenotype analysis. Some really cool analyses to find interesting biology e.g. allelic series and ancestry-enriched variants
18.09.2025 17:29 — 👍 0 🔁 0 💬 1 📌 0
A project many years in the process, we’re pleased to present our work on multi-ancestry meta-analysis across a boatload of traits in the UK Biobank: www.nature.com/articles/s41...
18.09.2025 17:25 — 👍 64 🔁 25 💬 1 📌 0We’ve put up summary statistics for over 3,000 traits in the All of Us resource, and a shiny new browser alongside it! Explore your favorite gene or phenotype here: allbyall.researchallofus.org #ASHG24
08.11.2024 20:32 — 👍 35 🔁 14 💬 1 📌 1Starter pack of people who create starter packs?
08.11.2024 00:06 — 👍 1 🔁 0 💬 0 📌 0You mean “ReNally???”?
07.11.2024 18:23 — 👍 0 🔁 0 💬 1 📌 0Heh, it was on our list but somehow never made it into the pre-submission checklist. Will do!
21.09.2024 17:00 — 👍 0 🔁 0 💬 0 📌 0Interesting question. We do have a “gnomAD-new” analysis in there but haven’t broken down by ancestry - i fear a lot is going to be driven by “not yet observed” (which is the same across all ancestries)
20.09.2024 16:45 — 👍 1 🔁 0 💬 0 📌 0It gets a bit more complicated though - these scores have a mix of impacts of variant-to-gene, as well as prioritizing which genes, when disrupted, lead to phenotypes. Perhaps a new method that combines both these insights optimally will outperform them all!
20.09.2024 15:51 — 👍 0 🔁 0 💬 1 📌 0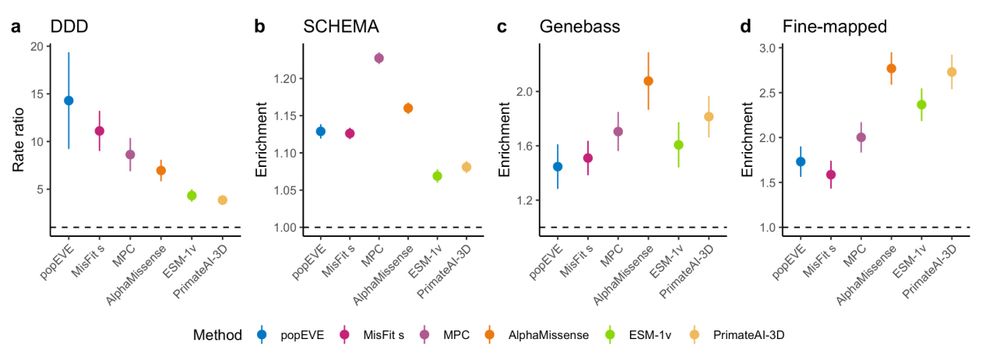
We found that population-focused methods do best for identifying highly impactful variants (de novo’s in individuals with developmental disorders for instance), while the deep learning methods are better at prioritizing inherited variation in biobanks
20.09.2024 15:49 — 👍 0 🔁 0 💬 1 📌 0
We have a new preprint that we’d love feedback on! We benchmarked a bunch of variant scoring methods to figure out what they were actually doing, and how they performed across selection regimes: www.biorxiv.org/content/10.1...
20.09.2024 15:47 — 👍 7 🔁 6 💬 2 📌 0Welcome new followers (and thanks @michelnivard.bsky.social)! I’m loving the critical mass, and to celebrate, I’ll post some exciting new content (my first time posting here and not on the the other site)
20.09.2024 15:46 — 👍 2 🔁 0 💬 0 📌 0
Recently out on #bioRxiv: our updated approach to identify regional variability in missense mutation intolerance (“constraint”) in protein-coding genes using the gnomAD database.
www.biorxiv.org/content/10.1...
1/10

As genomic analyses scale to millions of exomes/genomes, we need a scalable infrastructure to process/QC/handle these data while retaining all the metrics needed for downstream analysis. A new preprint from the Hail team proposes a way to do this! Comments welcome: www.biorxiv.org/content/10.1...
31.01.2024 15:40 — 👍 4 🔁 1 💬 1 📌 0Paella is good. With LOEUF I assumed the culmination would be an omelette but CHARR is better in a paella, so maybe it’s a multi-course meal
07.12.2023 18:10 — 👍 0 🔁 0 💬 1 📌 0Extended data figure 2b has exclusive exon-only. I think we internally made some with intermediate overlaps and it was an intermediate result as you’d expect
07.12.2023 18:09 — 👍 1 🔁 0 💬 0 📌 0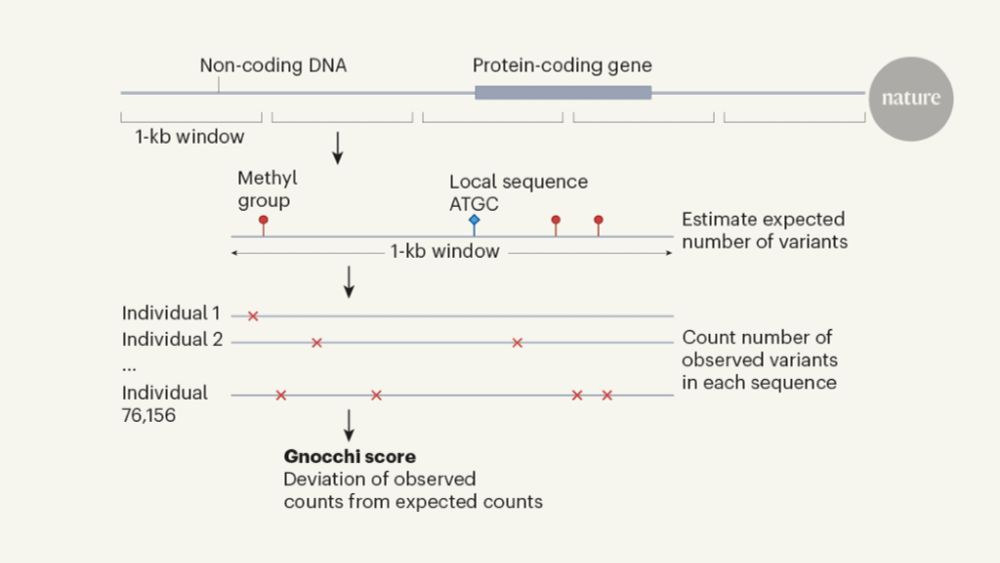
And thanks to Ryan Dhindsa and Slavé Petrovski for the excellent writeup and context around our work. Excited for the times ahead! www.nature.com/articles/d41...
06.12.2023 17:14 — 👍 2 🔁 0 💬 1 📌 0This is all thanks to an amazing production team, browser team, and steering committee @gnomad-project.bsky.social, the 76,156 individuals that provided their genomes, and support from Broad Genomics and Hail
06.12.2023 17:12 — 👍 1 🔁 0 💬 1 📌 0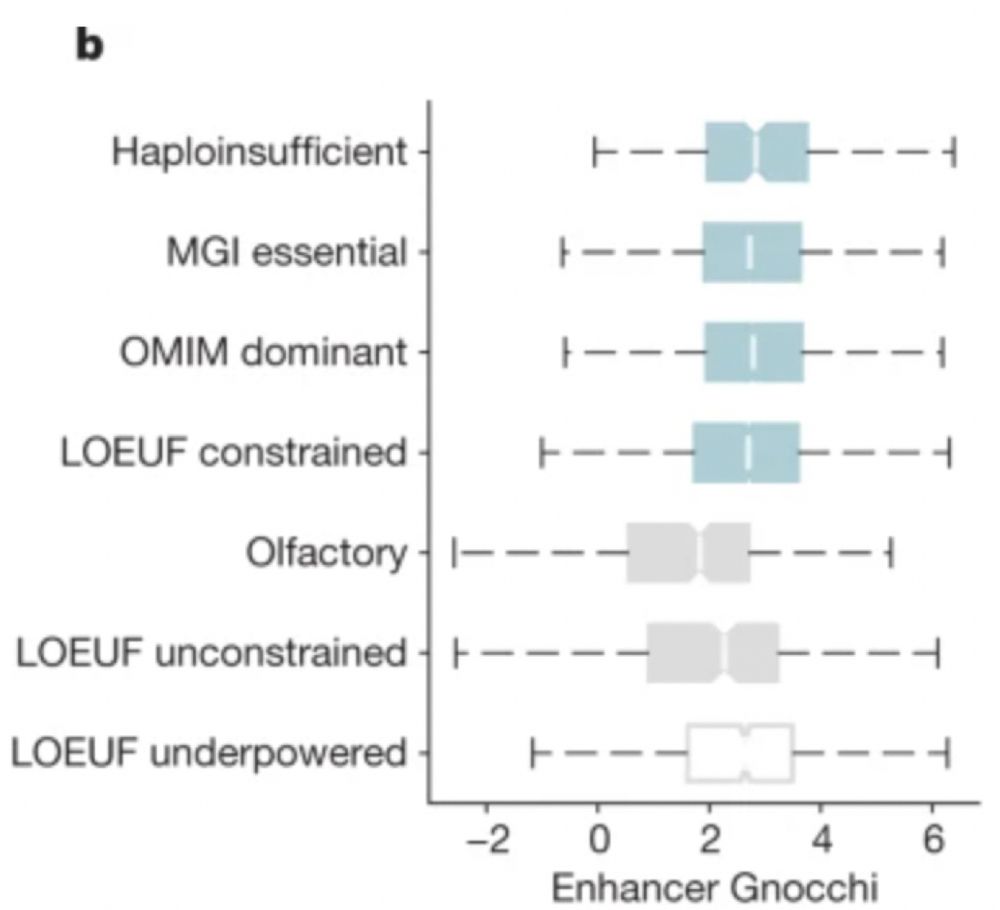
Interestingly, these scores also provide additional insight into genes regulated by these regions, even those underpowered by previous constraint metrics:
06.12.2023 17:11 — 👍 0 🔁 0 💬 1 📌 0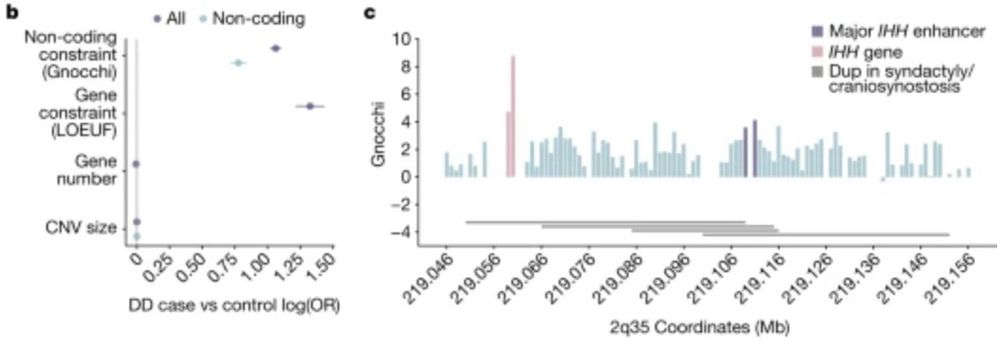
Gnocchi extends our constraint metrics to the non-coding genome, highlighting for instance, disease-associated non-coding CNVs
06.12.2023 17:11 — 👍 1 🔁 0 💬 1 📌 0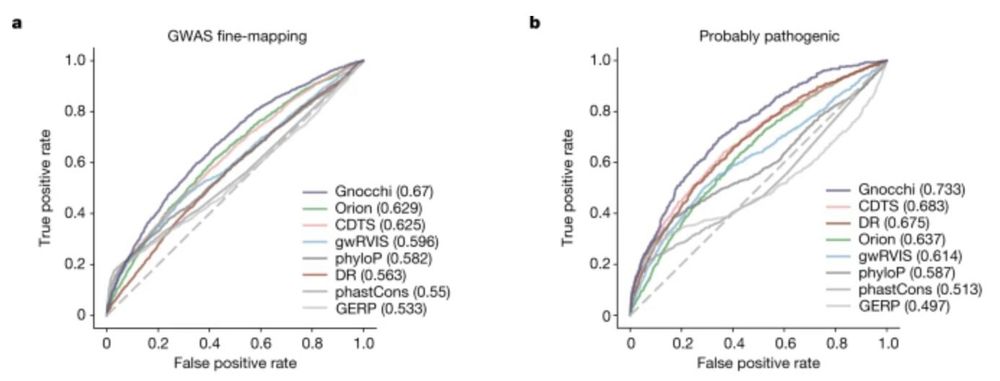
We built a new metric we called gnocchi (genomic non-coding constraint of haploinsufficient variation), building on methods that find depletions of variation (natural selection), which we show can prioritize functional variation
06.12.2023 17:10 — 👍 2 🔁 0 💬 1 📌 1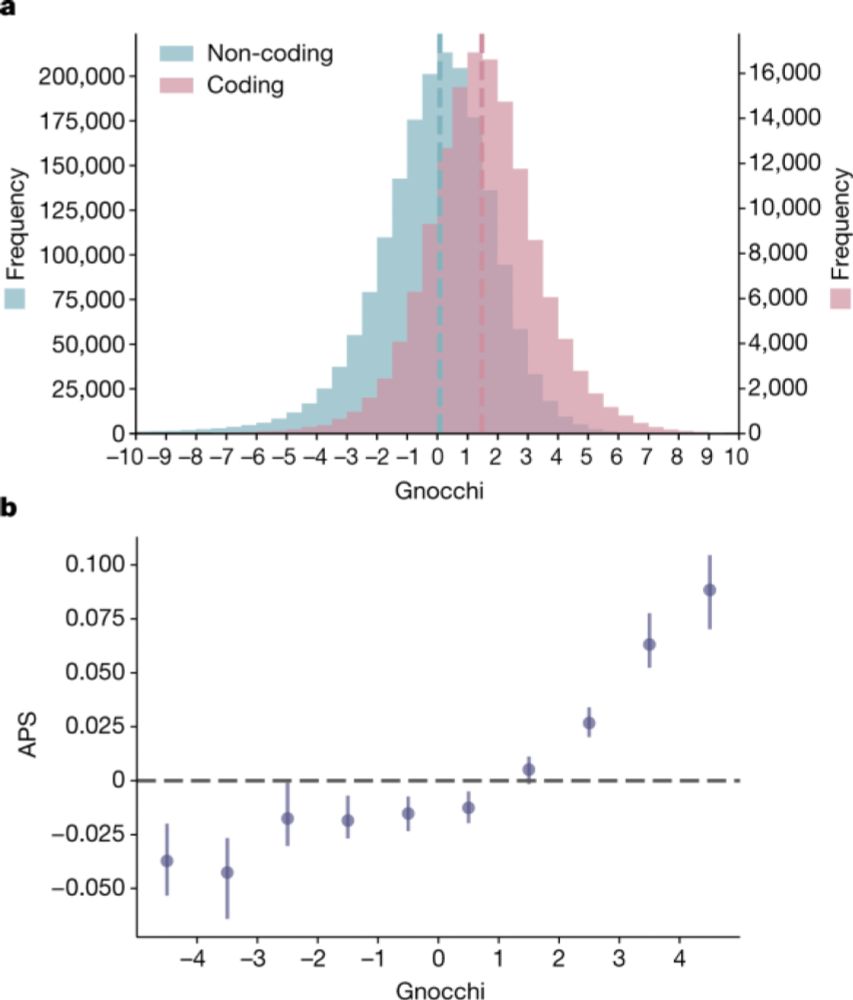
Thrilled to have our work on gnomAD out in print at Nature today. With 76K genomes, we can look beyond the coding genome and into the non-coding genome to find regions important for human disease idp.nature.com/authorize?re...
06.12.2023 17:10 — 👍 13 🔁 5 💬 1 📌 1Thanks to Wenhan Lu for driving this effort, Hail (hail.is) for building the scalable infrastructure that enabled this, and @gnomAD-project.bsky.social for the data and support
28.11.2023 19:36 — 👍 0 🔁 0 💬 0 📌 0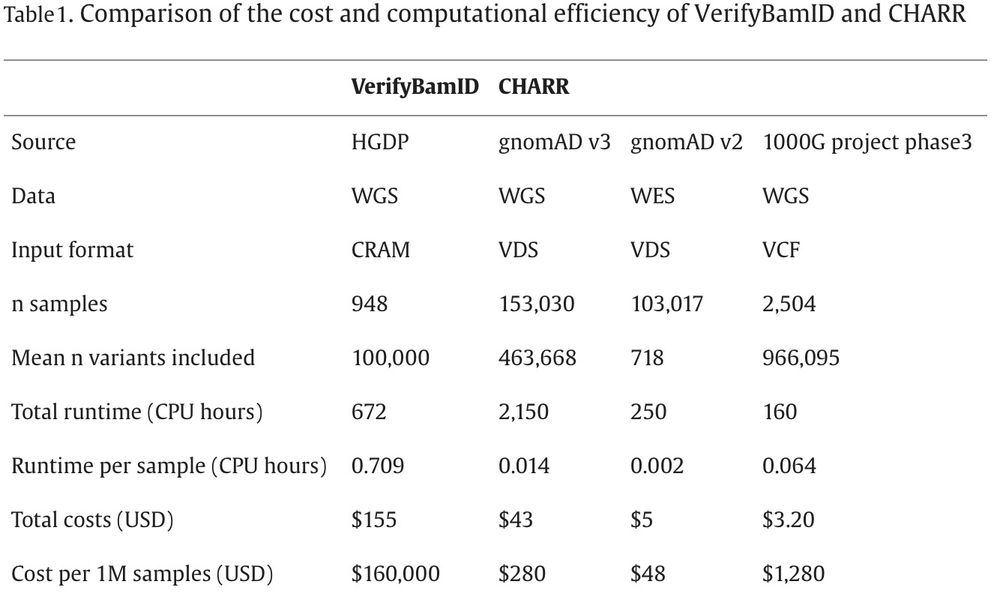
CHARR operates only on homozygous alternate sites and scales very well (“cost per 1M samples” might be my new favorite metric):
28.11.2023 19:35 — 👍 1 🔁 0 💬 1 📌 0Excited for my first post on this new site to share our work in print at AJHG using variant call data to estimate DNA contamination. As our sample sizes get into the millions of genomes, we need methods like this to efficiently process and quality control the data authors.elsevier.com/c/1i8PAgeX6LB~
28.11.2023 19:34 — 👍 12 🔁 4 💬 1 📌 0