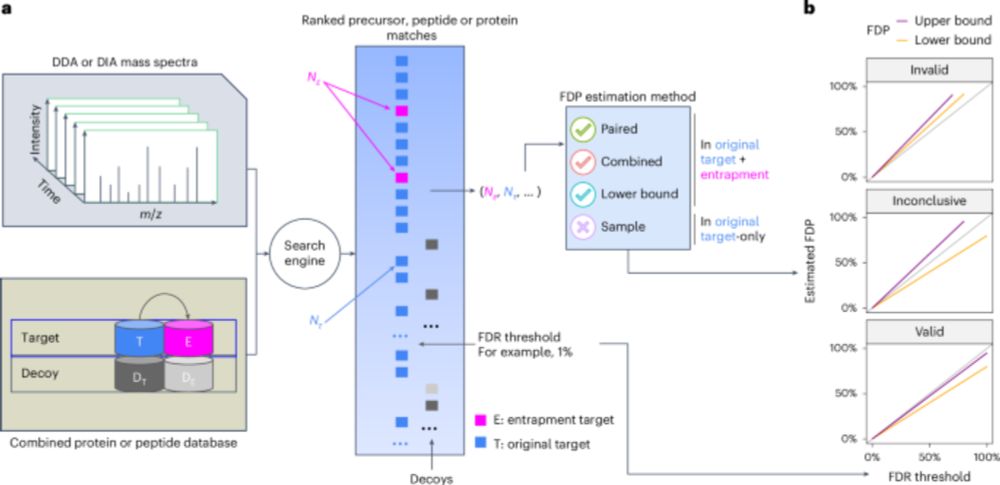
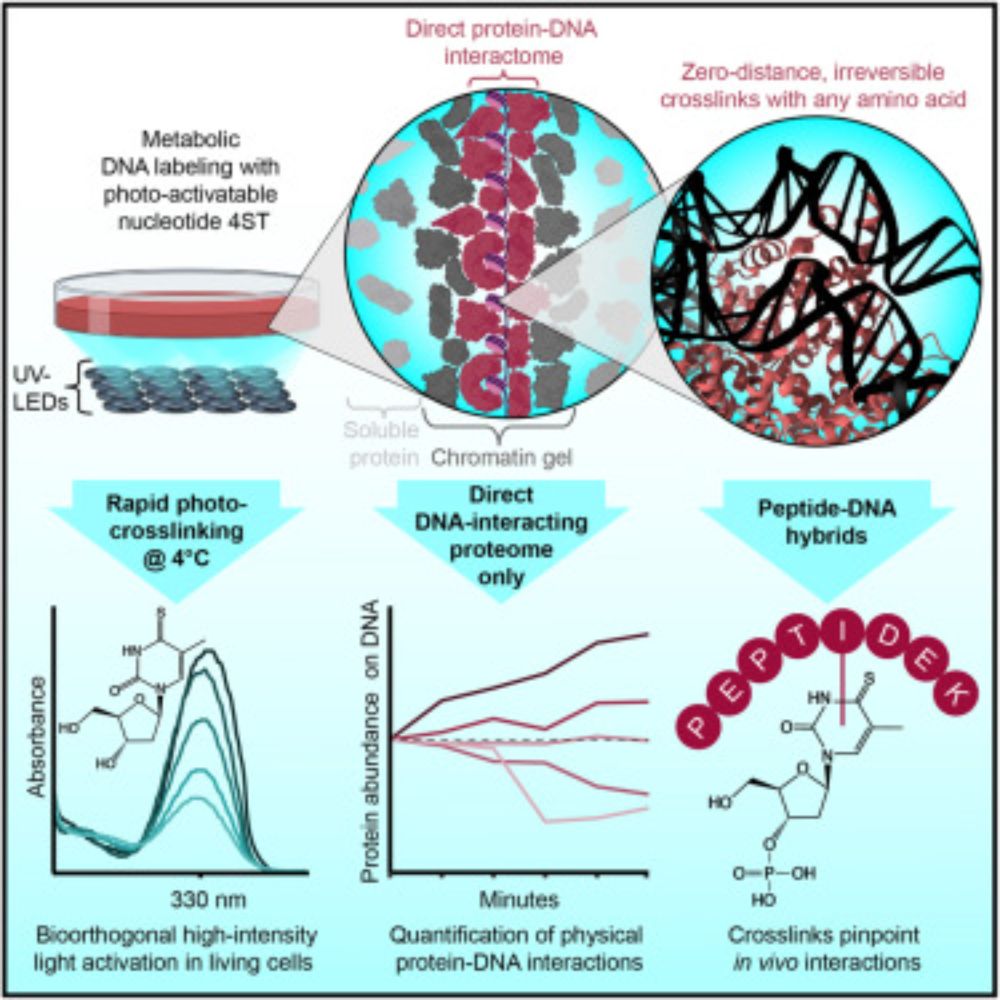
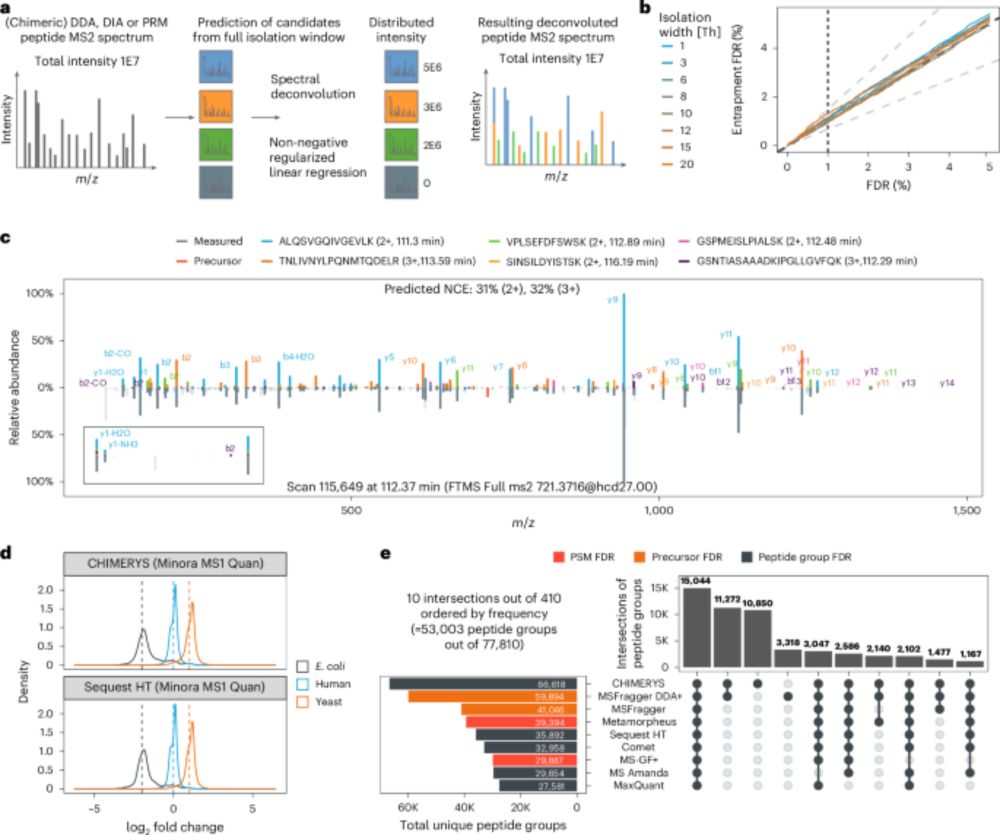
This is very useful.
Thanks for the effort !!
09.01.2026 17:56 — 👍 1 🔁 0 💬 0 📌 0

Proteoforms as the true units of physiological function link.springer.com/ar...
---
#proteomics #prot-paper
20.12.2025 10:00 — 👍 1 🔁 1 💬 0 📌 0
Yes. 100% agree.. That is bad MS2 co-isolation and ratio distortion :)
16.12.2025 15:41 — 👍 1 🔁 0 💬 0 📌 0
Good question how the *spectra* S/N is calculated by Thermo for the Astral...
16.12.2025 15:24 — 👍 1 🔁 0 💬 0 📌 0
One would need to multiple with MS1 intensity to get better quantities, because otherwise it depends when the TMT was picked how much intensity (ions/sec) you end up with in that moment of time, how many peptides you sum, etc... TMT is "only" excellent in conserving ratios within a TMT experiment.
16.12.2025 15:24 — 👍 1 🔁 0 💬 1 📌 0
I don't see *spectra* S/N ratios reported in table S3.
They have mean vs. STD *protein intensity* over three replicates. I didn't go deep enough to their analysis, but normally people just sum up TMT report intensity to protein level. This intensity is however not indicative of actual quantities.
16.12.2025 15:24 — 👍 0 🔁 0 💬 2 📌 0
my understating was that the S/N cutoffs are the sum over all TMT reporters. So a threshold of 1440 S/N for an 18plex would be average 80 S/N per reporter.. are the purple numbers retained spectra?
Thanks for sharing this analysis. Just sad that good signal leads to poor resolution in TOF world.
16.12.2025 13:24 — 👍 1 🔁 0 💬 1 📌 0
What I was always wondering is if you could do a normal ion processor/HCD-MS2 scan and then for the MS3 scan do first HCD in the IRM and then select N Fragments (by optics n=1, SPS in the IRM?) and do the second HCD in the processor?
But happy that you guys are thinking about it :) :)
20.11.2025 23:32 — 👍 2 🔁 0 💬 1 📌 0
(2/2) For me, the problem with the Astral rather is that it currently does not support MS3. I don't mind running the MS at a lower Hz if I can get much better data from it. Every time I meet someone from thermo, I try to spark the MS3 idea. It hasn't ignited yet ..
20.11.2025 14:50 — 👍 2 🔁 0 💬 1 📌 0
The Deu-TMT requires careful planning of which samples go to which channel and requires a Deu/nonDeu-"bridge" sample to remove the Deu-RT effect. Then it works as expected.
(1/2)
20.11.2025 14:50 — 👍 1 🔁 0 💬 1 📌 0
ProteomicsDB
A sneak preview is already online in proteomicsDB (www.proteomicsdb.org/analytics/KSR).
If you have questions, comments, or ideas, feel free to reach out.
19.11.2025 11:02 — 👍 1 🔁 0 💬 0 📌 0
In summary:
* 133 clinical kinase inhibitors x 5 cell lines
* >17 million peptidoform DRCs
* Critical re-evaluation of existing KSRs
* Proper FDR control for KSRs
* Identification of thousands of high-confidence KSRs
* Exposing how kinases shape cell line models and cancer patients
19.11.2025 10:44 — 👍 1 🔁 0 💬 0 📌 0

Chemical proteomics decrypts the kinases that shape the dynamic human phosphoproteome
Mass-spectrometry-based phosphoproteomics enables the analysis of thousands of protein phosphorylation events across the human proteome. However, there is a lack of scalable, hypothesis-free, and stat...
Did you ever come across a phosphosite in your proteomics data for which nothing was known? - I bet so!
We have developed a new strategy termed "potency coherence analysis" that leverages the drug potency dimension in decryptM to decode the kinases that shape the human phosphoproteome.
Read more:
19.11.2025 10:43 — 👍 25 🔁 9 💬 2 📌 1
The authors used CurveCurator to fit and evaluate all dose–response data across different omics assay types. CurveCurator’s scores let you set custom thresholds and interactively identify high-quality curves in ProteomicsDB.
18.11.2025 20:50 — 👍 0 🔁 0 💬 0 📌 0

Mapping drug mechanisms with ProteomicsDB: unified omics and cell sensitivity data at scale
Abstract. Proteomic and phenotypic cell sensitivity datasets are increasingly important for understanding chemoproteomics and the underlying drug mechanism
Very happy to see that dose-response curves are now super easy to access for everyone in proteomicsDB. Just a few clicks and you can see at which concentrations your favorite drug engages protein target(s), tinkers with signaling pathways, and inhibits cell growth. Read more: doi.org/10.1093/nar/...
18.11.2025 20:35 — 👍 6 🔁 1 💬 1 📌 0
A very clever approach to learn and predict MS2 spectra for modified peptides. By augmenting modification encodings and combining them with PROSIT, the new model has essentially generalized to ANY PTM - not just those in the training dataset. Super exciting !!
11.11.2025 17:14 — 👍 6 🔁 0 💬 0 📌 0
ProteomicsDB
Great to hear that you like it :)
To your question if that data is already in prDB - decryptM: YES it is ! e.g.
www.proteomicsdb.org/drug/107070/...
also i can recommend the interactive dashboards.html on zenodo to explore the data locally
zenodo.org/records/1609...
13.08.2025 16:38 — 👍 1 🔁 0 💬 0 📌 0
Very excited to see this story out in Science Signaling!!
Especially the use of dose-dependent profiling at different time points could clearly separate immediate from late and consequential signaling changes in KRAS-driven (phospho)proteomes.
31.07.2025 11:10 — 👍 3 🔁 0 💬 0 📌 0
Yes crazy... I still remember the 1.8 million PDF pages we were printing to manually determine what a real dose-response was in the original decryptM paper. Luckily, this is over now :)
Let me know if you need some tricks for the time-dependent hack and good luck with the paper
03.07.2025 13:25 — 👍 1 🔁 0 💬 0 📌 0
At the Kusterlab, we have developed a statistical analysis tool (CurveCurator) to mine these proteome-wide dose-response curves. You may want to give it a try. github.com/kusterlab/cu...
3/3
03.07.2025 12:37 — 👍 1 🔁 0 💬 1 📌 0
A 1.5 orders of magnitude dose range is not really wide enough for a full dose-response curve, but if you have such a scaled-up workflow, you could easily extend it to more doses with good dose resolution. 2/3
03.07.2025 12:36 — 👍 1 🔁 0 💬 1 📌 0
I really like the regulome-wide profiling in a dose-dependent fashion. Going one step further, I would love to see actual TF dose-response curves in Figure 5. So not interpreting each dose individually as bar, but actually have a curve with potency and efficacy estimates for each protein. 1/3
03.07.2025 12:36 — 👍 1 🔁 0 💬 1 📌 0
I am looking forward to discuss with with you:
• (phospho)proteome-wide dose-response profiling
• statistical analysis of 180 million curves with CurveCurator
• mapping kinase-resolved activities changes due to all target engagements
• (re-)evaluating the kinase substrate space in humans
04.06.2025 11:00 — 👍 4 🔁 0 💬 0 📌 0
Interesting.. that’s a long time for sure!
We are running the astral mostly without FAIMS and experienced for us normal exploris-like cleaning cycles.
08.04.2025 18:19 — 👍 2 🔁 0 💬 2 📌 0
It probably also means that a single publication by one group is not proving anything. Only if multiple independent groups report the same result it becomes trustworthy. Given the reproducibility crisis in biology, maybe this AI fabrication just accelerated a deeper problem, that we need to overcome
31.03.2025 18:52 — 👍 8 🔁 0 💬 1 📌 0
Old person forgetting everything they ever knew about proteomics at speeds in excess of 270 Hz...
Founder & CEO @jura.bsky.social | Full-stack probabilistic machine learning for the development of genetic medicines | NYC & Basel & Boston
Full-stack machine learning and synthetic biology company dedicated to transforming personalized, immune-mediated medicine
EMBO is the organization of more than 2,100 leading researchers that promotes excellence in life sciences in Europe and beyond.
https://www.embo.org/
Postdoc @ Küster lab | prev, PhD @ Trost lab | proteomics | chemoproteomics | mass spectrometry | target deconvolution | drug MoA
PhD student in proteomics & mass spectrometry @kusterlab
Postdoc at the University of Antwerp working on improving the identification rate of immunopeptides in TimsTOF data. Also interested in privacy and post-translational modifications.
Interested in Ubiquitin, Proteostasis and Signaling
Scientist at @mpibiochem.bsky.social
Alumnus of @mrclmb.bsky.social
MSAID transforms the way scientists analyze proteomics data
🌐 https://www.msaid.de
➡️ Request a Free CHIMERYS Demo: https://www.msaid.de/chimerys
Follow us: https://linktr.ee/msaid_de
#TeamProteomics #TeamMassSpec
Stem cell scientist interested in hematopoiesis and associated diseases. Single cell proteomics is also pretty cool.
We are the Savitski lab located @ EMBL using and developing proteomics methods for assessing the state of the proteome
Proteomics and metabolomics | Bacterial proteomics | opinions shared here are my own
Chemoproteomics @ Athens, Greece
#ChemBio
#chemoproteomics
https://orcid.org/0000-0002-4782-4029
Exploring biology with mass spectrometry
Professor @ Scripps Research | Department of Chemistry | chemical biology & chemical proteomics
A Reviews journal from SpringerNature. Timely Review articles, Comments and Highlights of the latest research in molecular cell biology. https://www.nature.com/nrm/
Research, news, and commentary from Nature, the international science journal. For daily science news, get Nature Briefing: https://go.nature.com/get-Nature-Briefing