Not to be lost - this is an assessment for discovery experiments to max. biological yield. Targeted will still benefit from added DPPP in many cases
31.01.2026 21:06 — 👍 1 🔁 0 💬 0 📌 0
I think the reality is that the model in 2020 paper is an ideal scenario for uniform high SNR peptides. In reality, ideal isn't probable for all acquisition. Greater divergence decreases DPPP : accuracy dependency in model. Low SNR is still very much dependent on DPPP to enable LOQ assessment.
31.01.2026 18:20 — 👍 1 🔁 0 💬 1 📌 0
Yep! Can recreate the plot, but added detector characteristic, SNR, and peak asymmetry. Esp. SNR impacts the quant. If I drop SNR from the model it looks basically identical to the 2020 MCP paper.
31.01.2026 18:17 — 👍 1 🔁 0 💬 1 📌 0
Check out my US HUPO poster and soon to come pre-print :)
Good chromatography and high SNR peaks will be fine with few DPPP. Low abundance features suffer with few DPPP.
29.01.2026 22:09 — 👍 3 🔁 0 💬 2 📌 0
Discord chat room?
05.12.2025 22:11 — 👍 1 🔁 0 💬 0 📌 0
I'm excited to share new results at #HUPO2025!
I’ll be presenting our latest work on a next-generation DIA search and FDR pipeline that enables sensitive, accurate and scalable proteomic analysis — in just a fraction of the time required by current algorithms.
📍 Poster PV.01.009 — Monday
08.11.2025 12:28 — 👍 10 🔁 1 💬 0 📌 0
Clinical protocols are very tricky to standardize across sites.
14.09.2025 12:43 — 👍 4 🔁 0 💬 1 📌 0

Pre-analytical drivers of bias in bead-enriched plasma proteomics | EMBO Molecular Medicine www.embopress.org/do...
---
#proteomics #prot-paper
13.09.2025 08:40 — 👍 13 🔁 5 💬 2 📌 0
Yeah just run a sample every 2 minutes for 5 years with no overhead or down time and a 2M instrument costs 1.5 per sample…
Pretend 15 min with 75% up time and it’s 15/sample. Adjust cost relative to purchase price.
03.08.2025 14:42 — 👍 1 🔁 0 💬 0 📌 0
Isomerization? Especially in deamidation prone peptides with NQ. This does split peaks in LC. Eye lens work has seen this for decades. Doesn’t mean deamidation necessarily. Can generate 4plet or more if including modification. Tris in prep?
08.07.2025 20:49 — 👍 1 🔁 0 💬 0 📌 0
Or on high mass? I haven’t seen much for >100ng from the 8600.
28.06.2025 14:41 — 👍 1 🔁 0 💬 1 📌 0
Not that I’m aware of - though I’d also be nervous about charge capacity on transmission through the tims device. I’d rather see the 7600 or 8600.
28.06.2025 13:17 — 👍 1 🔁 0 💬 0 📌 0
It’s unclear that SLIMS has the reproducibility or resolution to achieve this. Marketing figures aside, peptides don’t always make nice Gaussians. Co-resolving charge states likely isn’t an issue.
What I want to see is an honest effort on DIA vs PAMAF with a good DIA TOF instrument.
28.06.2025 12:32 — 👍 3 🔁 0 💬 1 📌 0
Rationale seems to be more ions = better data. Astral transmits ~1/200th of ions within a target search space at a time (less the overhead and MS1 times).
A 400ms SLIMS separation could reasonably replace half the selectivity of the quad and 4x throughput of qTOF.
28.06.2025 12:30 — 👍 1 🔁 0 💬 1 📌 0
When I check in every month or so, it’s dry. Much drier than bsky
15.06.2025 13:38 — 👍 4 🔁 0 💬 0 📌 0
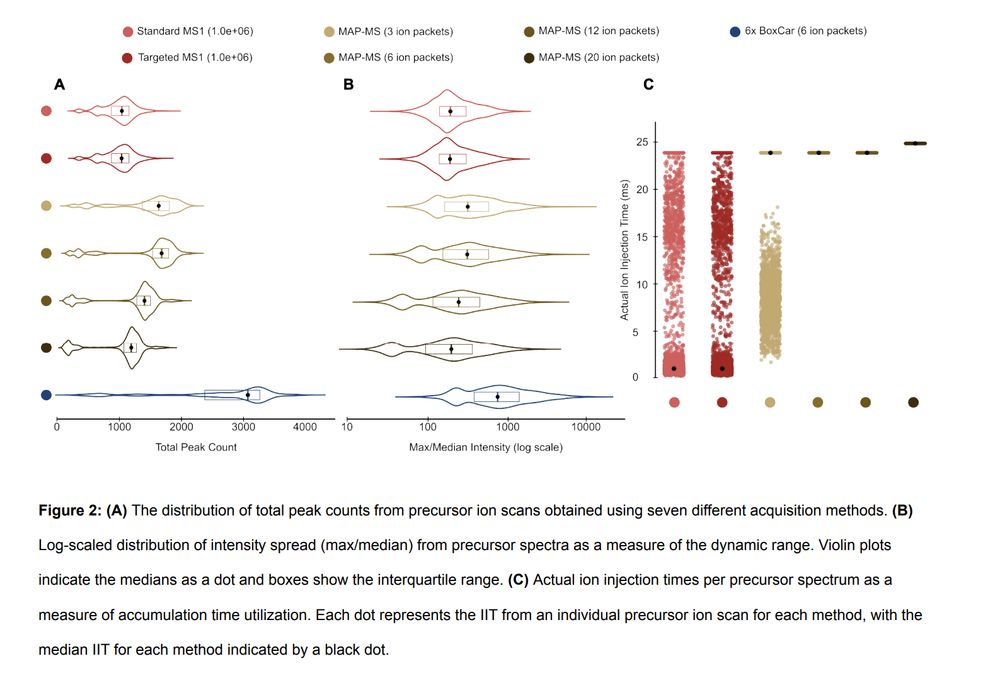
Improving proteomic dynamic range with Multiple Accumulation Precursor Mass Spectrometry (MAP-MS) www.biorxiv.org/cont...
---
#proteomics #prot-preprint
18.05.2025 15:40 — 👍 9 🔁 2 💬 0 📌 0
For the right biological question, low IDs may even be preferential if they are subcomponent oriented. I'd argue that any method has its biases, both towards abundance and structural subcomponent.
10.04.2025 15:05 — 👍 0 🔁 0 💬 0 📌 0
However, the readout really shows that the sample was not rich in the target subpopulation for that method.
Toolset comparisons should really focus on the same plasma preparation protocol. Ideally there would be robust discussion to the tradeoffs of protocol to implemented toolset comparisons.
10.04.2025 15:00 — 👍 0 🔁 0 💬 1 📌 0
Implementation of any toolset should be done consistently within a project, but comparing study a to study b with different clinical protocols is a bit challenging.
Plenty of techniques now do well for EV proteins. But a double spun sample will typically be low EV. Readout is bad IDs.
10.04.2025 14:58 — 👍 0 🔁 0 💬 1 📌 0
There's huge dependency on the sample and its consistency in preparation for observables... Double spun plasma is very distinct from single-spun. Storage conditions and a myriad of other variables also impact readouts.
Plasma proteomics is ultimately a tool for clinical sciences to implement.
10.04.2025 14:56 — 👍 0 🔁 0 💬 1 📌 0
Not fully following. But you’ll have MS data files and search result files depending on parameters. There are wrong ways to search, but not a single right way. Upload ideally includes both data and search files.
A 500 sample study can easily exceed TB data size on most any MS.
07.04.2025 00:07 — 👍 1 🔁 0 💬 0 📌 0
Loading 1k astral files searched by Diann in a R interface is relatively painful in my opinion. There’s probably more necessary innovation to search, process and share data from mega cohorts that are now arriving.
06.04.2025 12:36 — 👍 2 🔁 0 💬 0 📌 0
Unfortunately, MS data is massive. Many observations in a vector multiplexed by LC. New instruments have been “worse” about this.
OLink is quite smart with data presentation being a neat and contained parquet.
06.04.2025 12:32 — 👍 2 🔁 0 💬 1 📌 0
I’m very pro data sharing. I’ve benefited a number of times from it and been limited by it a number of times. Even available upon request or application data may as well be not shared.
Maybe a challenge is upload. I doubt MOMI 46k astral bio samples will be shared. Well over 1 PB…
06.04.2025 12:29 — 👍 2 🔁 0 💬 2 📌 0
Disagree. There can be legal or IP reasons to not share. Probably why a lot of expensive OLink data wasn’t shared, before. IP can be mined by anyone once out there.
Also human data sharing issues etc.
I wish at least control files would be shared at minimum to validate functionality, though.
06.04.2025 12:27 — 👍 2 🔁 0 💬 3 📌 0
“chemical and physical biology; structural biology track”
Realistically I was housed in biochemistry and chemistry. Vanderbilt fortunately has two dedicated MS courses, one specifically in proteomics. Another class for using SIMION with John McLean too.
23.03.2025 22:50 — 👍 1 🔁 0 💬 0 📌 0
In grad school - 2018
21.03.2025 12:58 — 👍 1 🔁 0 💬 1 📌 0
Official Account for Christina Woo’s Lab at Harvard CCB| chembio, protein mods, chemoproteomics, TPD | Student-run account | posts by Christina signed CMW
Protein Mass Spectrometry
Emmott Lab | CPR | University of Liverpool
Protein complexes and malaria parasites in #ZBlab @NTUsg. Previously PhD at @EMBL Loew lab. https://github.com/sampazicky
Scientist @UniMelb 🇦🇺 focusing on #proteomics, signaling and metabolism. Loves 🏄♂️⛷️⛰️
A mass spec proteomics user in the CAR-T therapeutic space
Proteomes (ISSN 2227-7382) is an #openaccess, peer reviewed journal on all aspects of #Proteomics science, published quarterly online by @mdpiopenaccess.bsky.social
Postdoc @ Küster lab | prev, PhD @ Trost lab | proteomics | chemoproteomics | mass spectrometry | target deconvolution | drug MoA
Leading provider of antibody reactome services. Catalog antigen libraries include human proteome, virome & allergome. Custom library creation also possible!
Check us out: https://www.infinitybio.com/
The Human Proteome Organization (HUPO) is an international scientific organization representing and promoting proteomics through international cooperation and collaborations
Absea Biotechnology | Empowering protein science
14,000+ recombinant proteins & 11,000+ monoclonal antibodies covering 70% of the human proteome.
Germany-USA-China
https://www.absea.bio/index.html
Delivering on the Promise of the Human Proteome
MOBILion’s tech separates and analyzes challenging molecules with higher resolution, faster analysis, and simpler workflows—advancing safer, effective therapies
Mass spectrometry, metabolomics, lipidomics. Wannabe cyclist. Views are my own and not my employer’s.
Founder of GoldenHaystack Lab (www.goldenhaystack.org)
Interested in proteomics technologies, pQTLs, proteome variation in health and diseases. Views are my own.
The Mann Lab is a pioneer in mass spectrometry-based proteomics. Posts represent personal views from lab members and Matthias Mann