

Cricket and Willow hosted a W-S lab dinner!
15.09.2025 13:40 — 👍 4 🔁 1 💬 0 📌 0
@hkws.bsky.social
Avid rower who sometimes thinks about biomolecular dynamics. Asst Prof @uwbiochem.bsky.social https://waymentsteelelab.org/
Cricket and Willow hosted a W-S lab dinner!
15.09.2025 13:40 — 👍 4 🔁 1 💬 0 📌 0
The W-S lab is seeking to hire a research scientist!
This person will lead wet-lab innovations in our team as we combine AI and experiments to understand and design dynamics!
Thanks for sharing/RTs!
jobs.wisc.edu/jobs/scienti...

We are looking to hire (yes, even in this economy!) a jr. specialist to train in protein prep/structural biology related to our AVOID-ome work as part of openadmet.org.
A great position for someone who is looking to be a tech for a few years before grad or med school.
aprecruit.ucsf.edu/JPF05424
Join us for the 2nd Macromolecular Conformational Ensembles Conference on June 9th/10th at UCSF. The most compelling questions in structural biology cannot be effectively addressed using only a single structure. conformationalensembles.github.io @fraserlab.bsky.social
31.03.2025 13:41 — 👍 22 🔁 13 💬 2 📌 2someone said we need a meme
12.05.2025 15:35 — 👍 27 🔁 6 💬 1 📌 0
btw in the name of getting NMR dynamics data out there, RelaxDB is available at the Dyna-1 huggingface! Go check it out huggingface.co/gelnesr/Dyna-1
09.05.2025 21:08 — 👍 6 🔁 1 💬 0 📌 0
Did some quick curation for ~30 of 133 proteins in our new dataset RelaxDB, which is the first of its kind to gather experimental info on timescales of motions per residue. The x-axis is intentionally silly to make a point - these dynamics expts do not use more than 10% D2O in samples.
09.05.2025 21:07 — 👍 4 🔁 0 💬 1 📌 0@dereklowe.bsky.social The paper you cite discusses difference when entire solvent is D2O. It could be that D2O partitions away from protein when it’s there at 5% since the paper’s main point is H2O has increased interactions with proteins. Curious if anyone’s investigated that!
09.05.2025 19:22 — 👍 12 🔁 1 💬 1 📌 0this is why many protein nmr spectroscopists only use 5-10% D2O! I hope this doesn’t deter people from what imo is a super undervalued source of data
09.05.2025 19:03 — 👍 8 🔁 1 💬 1 📌 0👏 models in nmr structure != thermodynamics 👏
Structures I’ve been part of include the top-X most probable structures. Same as how the top-X-scoring models from a Rosetta run wouldn’t be a thermodynamic ensemble.
If we wanna make accurate boltzmann samplers, we gotta know what distributions they should be sampling 😁 These change in the presence/absence of ligands, and there are multiple systems for which this is well-understood by this point experimentally.
28.04.2025 12:59 — 👍 9 🔁 1 💬 0 📌 0
BioEmu actually doesn't pass this test for AdK: majority of samples are in closed state (1AKE), which is the same intrinsic bias that AF2 + random sampling gets.
I don't know the other apo/holo systems bioEmu looks at as well, but same story: sampled density is primarily at the holo state.

You can see this in this FRET data for AdK here: in ligand-free form, the open state is more populated than closed.
28.04.2025 12:59 — 👍 0 🔁 0 💬 1 📌 0Really nice resource from @delalamo.xyz !!
Wanna mention the "AdK test" we're noticing newer DL methods fall short on:
many proteins (like AdK) that have apo/holo conf change sample both the apo/holo state even without ligand bound. But w/o ligand bound, they *are mainly in the apo state*

Next Tues (4/29) at **4:30PM** ET, we will have @ginaelnesr.bsky.social @hkws.bsky.social present "Learning millisecond protein dynamics from what is missing in NMR spectra"
Paper: biorxiv.org/content/10.1...
Sign up on our website for zoom links!
Your yearly reminder to acknowledge the core facilities you use and their staff scientists in your papers. These scientists are a crucial part of the scientific ecosystem and to continue to exist they need tangible credit for their work. Plus their associated expertise adds credibility to your work.
10.04.2025 13:38 — 👍 488 🔁 165 💬 7 📌 16I am sure future improvements exist over what we did! We removed deuterated samples (incomplete back-exchange) and entries with more than 12 15N assignments missing in a row. This is why we call this a "bold assumption", to me the proof is in the pudding, that we got a model with signal at all!
21.03.2025 19:35 — 👍 3 🔁 0 💬 1 📌 0Hi Gabe! Yeah we thought of af pair (no MSA) as upper limit but what you’re proposing would also be control for pair rep.
To me, the kicker things we want to predict are coordinated motions that have low prob. Saw hints of that with CypA. I don’t think people would think of that as flexibility …




Unassigned nitrogens in nmr data often indicate biologically relevant motion in proteins, and this can be used train deep learning models of protein dynamics!
Hannah Wayment-Steele
@ginaelnesr.bsky.social @sokrypton.org
www.biorxiv.org/content/10.1...
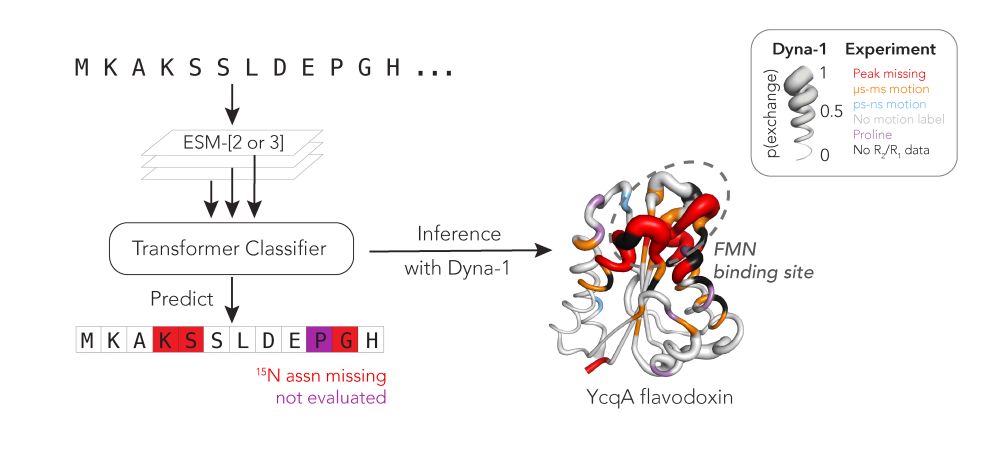
Protein function often depends on protein dynamics. To design proteins that function like natural ones, how do we predict their dynamics?
@hkws.bsky.social and I are thrilled to share the first big, experimental datasets on protein dynamics and our new model: Dyna-1!
🧵
Thank you so much to the amazing Doro Kern for dreaming big w me. Thank you to @ramith.fyi, Hasindu, and @sokrypton.org for pushing these ideas in early days! last but not least, thanks to @jcchildsfund.bsky.social and @hhmi.org for funding :))
20.03.2025 15:02 — 👍 4 🔁 1 💬 1 📌 0Tremendous thank you to partner-in-crime @ginaelnesr.bsky.social. This collaboration started when she offered to clean a metadata spreadsheet, and she ended up pushing the deep learning we tested so much further than I could have alone!
20.03.2025 15:02 — 👍 4 🔁 0 💬 1 📌 0
Moral of the story: useful data is out there at many degrees of quality, but we need to know how to interpret the data. We're so excited to see where these models and data go next!
Paper: rb.gy/de5axp
Dyna-1 colab (thanks to @ginaelnesr.bsky.social ) and RelaxDB: github.com/WaymentSteel...
Dyna-1 has predictive power in the big dogs of dynamics experiments: CPMG relaxation-dispersion. Dyna-1 predicted high p(exchange) in some aa's that typical data treatment says has no Rex, but more careful consideration says is real (NMR aficionados: unsuppressed R2).
20.03.2025 15:02 — 👍 3 🔁 0 💬 1 📌 0
There are so many interesting things that Dyna-1 predicts, but I wanna talk about a trend it didn't predict in RelaxDB! We realized that in many RelaxDB datasets where Dyna-1 did poorly, the Rex came from phosphate buffer binding/unbinding to the protein, an experimental artefact.
20.03.2025 15:02 — 👍 3 🔁 0 💬 1 📌 0
We split the mBMRB into train, val, and test set, and held out RelaxDB too as eval set. Sure enough, many pre-trained models that we tried - AF2, ESM2, ESM3 - had predictive power for Rex in RelaxDB. The best model we found was a middle layer of ESM3. We named it Dyna-1. Here's NtrC from test set:
20.03.2025 15:02 — 👍 4 🔁 0 💬 1 📌 0We made the bold assumption that missing assignments were aa's with µs-ms motion. If we could train a model that could predict those, would it have learned µs-ms motion?
20.03.2025 15:02 — 👍 3 🔁 0 💬 1 📌 0I next curated the "missing BMRB": 9,381 proteins with the aa labels: 0: 15N assn is present, 1: assn is missing. These labels are quite distinct from other properties like b-factors and missing residues in X-ray and EM (see paper for more).
20.03.2025 15:02 — 👍 3 🔁 0 💬 1 📌 0
Some aa's in RelaxDB didn't have Rex data bc they were missing from assignments, step 1 of any NMR study. This can happen if a peak is exchange-broadened due to µs-ms motion.
AA's missing assns were also conserved.
Then I realized - we have that data for >10,000 proteins, way more than 133!

I found a trend that amazed me in RelaxDB: aa's with µs-ms motion were more conserved than aa's with no motion, which were more conserved than aa's with ps-ns motion.
**Depending on what timescale of motion we want to predict, we should expect different evol patterns.**
What's the red category?



















