Very exciting! I tried out the inference code but the script is current crashing after conformer generation due to missing the AimNet model weights. Did I miss a download link somewhere?
21.08.2025 00:41 — 👍 0 🔁 0 💬 1 📌 0

GitHub - isayevlab/LoQI: LoQI: Low Energy QM Informed Conformer Generation
LoQI: Low Energy QM Informed Conformer Generation. Contribute to isayevlab/LoQI development by creating an account on GitHub.
The paper represents a paradigm shift by combining machine-learned interatomic potentials (MLIPs) with generative modeling to bypass traditional conformer generation, achieving both higher accuracy and greater efficiency than existing methods. Free & open source: github.com/isayevlab/LoQI
20.08.2025 16:45 — 👍 7 🔁 1 💬 1 📌 0
Interested in doing a postdoc at DFCI/Harvard on computationally designing and experimentally characterizing mini-protein binders for biomedical applications? Eric Fischer and I are looking for someone to work in our groups starting asap! Email me or my admin with a CV to apply!
19.08.2025 16:21 — 👍 3 🔁 2 💬 0 📌 0

🚨New paper 🚨
Can protein language models help us fight viral outbreaks? Not yet. Here’s why 🧵👇
1/12
17.08.2025 03:42 — 👍 42 🔁 19 💬 3 📌 0

Hardware/wetware codesigned data loop VISTA makes use of generative model sampling and synthesis "on chip" on-board by leveraging oligosynthesis setup shown here.
The biggest challenge for AI in biology isn't just models, it's the data used to train them. Standard biological data isn't built for AI. To unlock generative AI for drug discovery, we must rethink how we generate and capture data. 1/
22.07.2025 12:29 — 👍 29 🔁 9 💬 2 📌 6
Interesting though that Boltz-2 ipTM seems to rank the affinities /almost perfectly for our point mutants and SAR. Curious to see what others are using to rank designed interfaces 🤔 2/2
07.06.2025 00:22 — 👍 1 🔁 0 💬 0 📌 0

Trying out the Boltz-2 affinity prediction on the Exatecan binders we generated with LASErMPNN and NISE. Affinity prediction still clearly has room to improve, but the model seems to be able to identify the highest affinity mutant in this small dataset. Thanks to @gcorso.bsky.social and team! 1/2
07.06.2025 00:22 — 👍 7 🔁 1 💬 1 📌 0

from most recent Harvard lawsuit. sums it up pretty succinctly I think
27.05.2025 14:36 — 👍 3 🔁 1 💬 0 📌 0
Congrats Liana 🎉
24.05.2025 19:24 — 👍 2 🔁 0 💬 0 📌 0
YouTube video by Boston Protein Design and Modeling Club
Design of small molecule binding proteins using deep learning
in case you missed the superb seminar that Ben Fry gave back in January, you can now check out the recording youtu.be/IgFgAYQrke4
and the preprint www.biorxiv.org/content/10.1...
03.05.2025 14:07 — 👍 2 🔁 3 💬 0 📌 0
We're super excited by the method. We think it can help to rapidly produce binders to small molecules for sensors, antidotes, delivery vehicles, even enzymes. Let us know what you think and please try it out! Finally, shout out to Ben and Kaia for making this all happen!!! 🤩
28.04.2025 15:22 — 👍 2 🔁 1 💬 1 📌 0

absorbance data showing that epic protect exatecan from hydrolysis
Lastly, Kaia checked to see if EPIC and its higher affinity mutant are able to protect exatecan from hydrolysis, which is not something serum albumin can do. For a drug that normally hydrolyzes in a few hours, EPIC was able to stabilize the lactone form for days! ✅
28.04.2025 15:22 — 👍 1 🔁 1 💬 1 📌 0

co-structure predictors agree with crystal structure but differ at ligand
Since EPIC and exatecan aren't in the PDB, we wanted to see how co-structure predictors do on it. They each get the backbone right but differ at the ligand. The pose is correct but the modeling of the conformer is wonky. AF3 does the best. AF3 is also able to rank affinities via pLDDT of ligand! 😱
28.04.2025 15:22 — 👍 2 🔁 1 💬 1 📌 0

crystal structure of EPIC agrees with design
Kaia was able to crystalize EPIC and determine its structure to 2.0 Å resolution. It agreed pretty well with the LASEr design! RFAA had a hard time modeling the lactone ring of the drug, so there is some disagreement there. The lactone is buried as intended, and the goal was to hide it from water 👍
28.04.2025 15:22 — 👍 2 🔁 1 💬 1 📌 0

example of neural proofreading to improve affinity
Ben didn't stop there. He wanted to improve affinity of EPIC for exatecan using computation alone. He used LASErMPNN to "proofread" EPIC's sequence using a predicted co-structure as input. LASEr suggested two mutations. Kaia verified that each improved binding 10x. 100x when combined (1 nM Kd)!
28.04.2025 15:22 — 👍 1 🔁 1 💬 1 📌 0
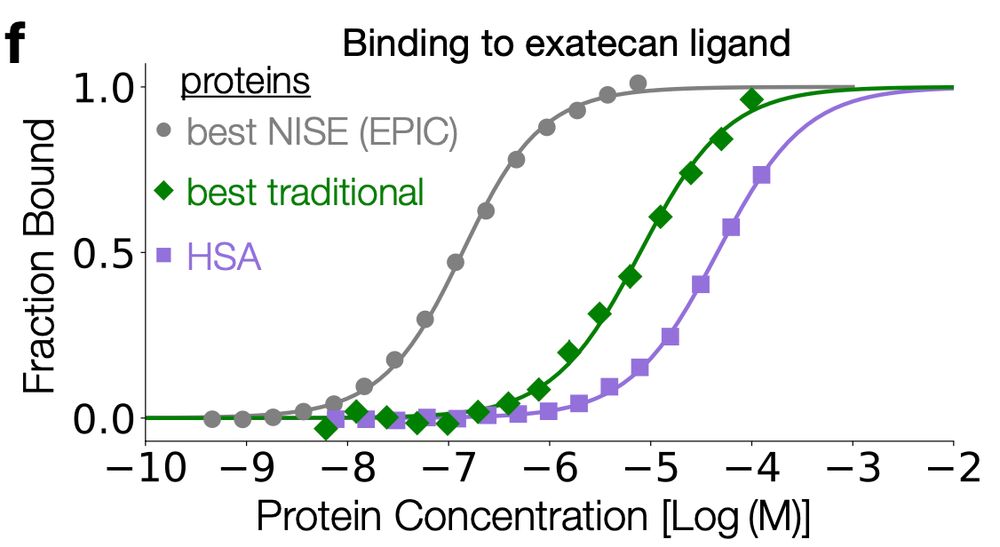
binding curves
Kaia Slaw (no bluesky) experimentally tested 4 designs from NISE and 16 from COMBS. All 4 NISE designs bound! The highest affinity binder- which Ben and Kaia call "EPIC" - was pretty tight (0.1 uM Kd). Compared to COMBS (3 of 16 bound, tightest was 10 uM), NISE and LASErMPNN did a much better job!
28.04.2025 15:22 — 👍 1 🔁 1 💬 1 📌 0
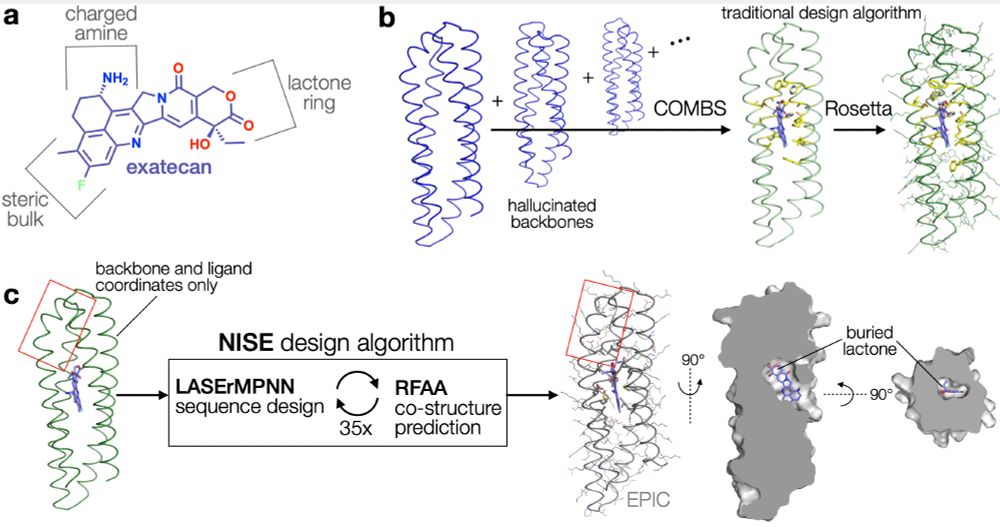
image of exatecan drug and design pipelines using COMBS and NISE
Ben used NISE and LASErMPNN to design binders to exatecan, an anticancer drug prone to inactivation by hydrolysis. We also used a more "traditional" approach using COMBS and Rosetta to design binders. We could compare the methods head to head.
28.04.2025 15:22 — 👍 1 🔁 1 💬 1 📌 0
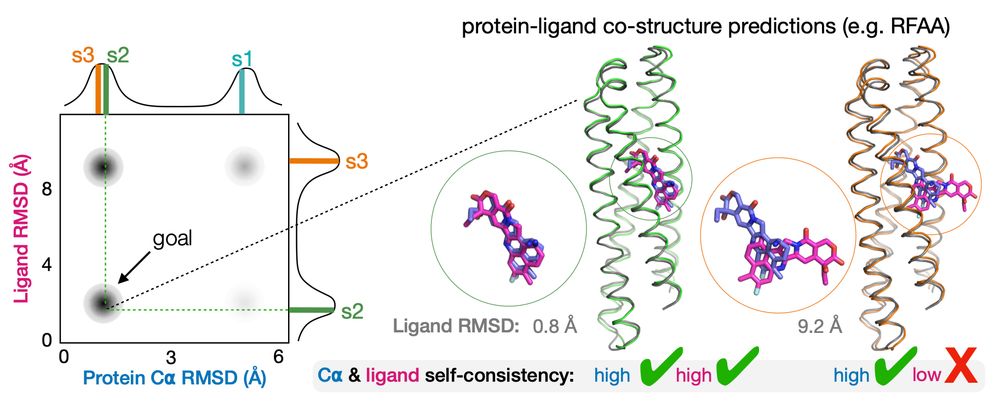
two proteins that have good predicted structures but only one has a self consistent ligand position
With the new co-structure predictors like RFAA, Boltz-1, and AF3, we can now extend self-consistency into the ligand dimension. And Ben's NISE algorithm maximizes this. Code repo here: github.com/polizzilab/N...
28.04.2025 15:22 — 👍 1 🔁 1 💬 1 📌 0
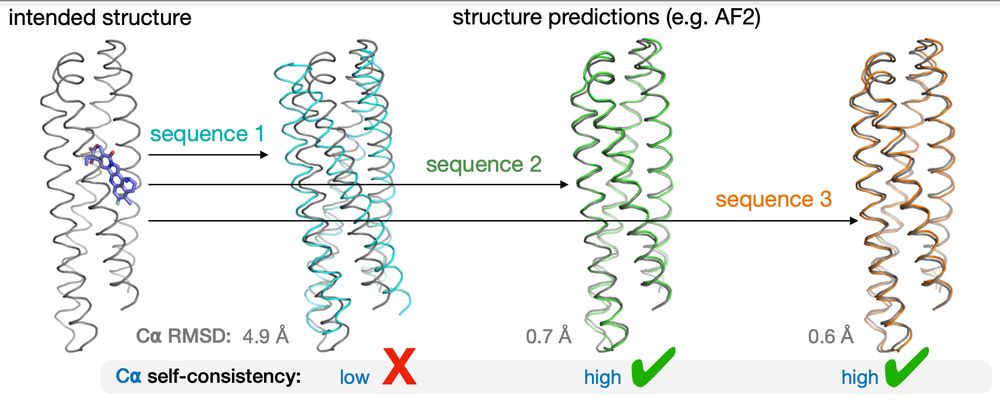
protein sequence design and structure prediction showing some designs agree with the intended structure
We all know in protein design about the goal of self consistency. That is, we want the predicted structure to look like the structure for which we designed the sequence.
28.04.2025 15:22 — 👍 1 🔁 1 💬 1 📌 0
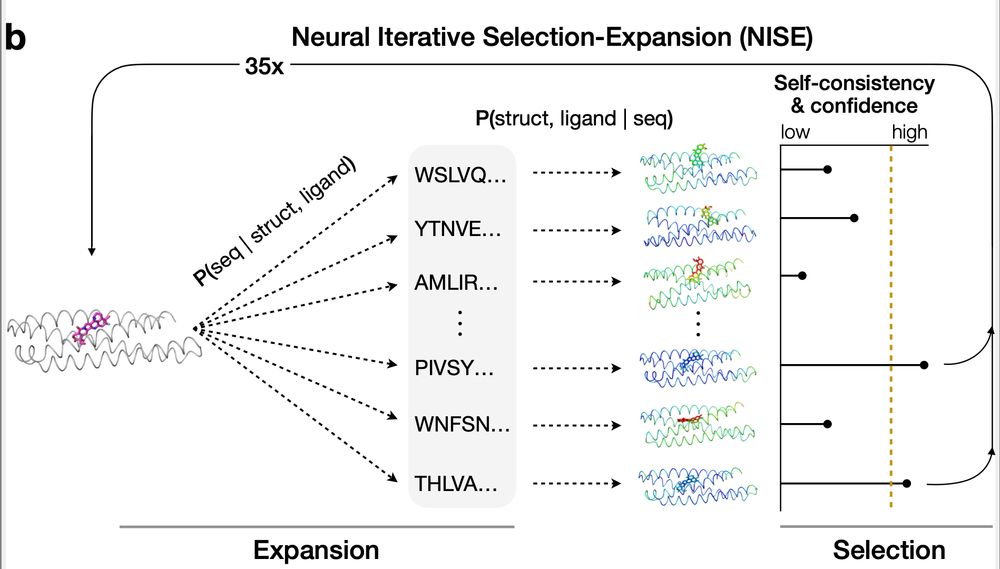
the NISE design algorithm iteratively optimizes a starting model through many rounds of sequence design and structure prediction
Ben used LASErMPNN in combination with a protein-ligand co-structure predictor, RFAA, in an iterative algorithm called NISE that refines designs. NISE optimizes the sequence, structure, and ligand conformer together to improve the confidence of both models. It's a neural-network-only algorithm
28.04.2025 15:22 — 👍 2 🔁 1 💬 1 📌 0
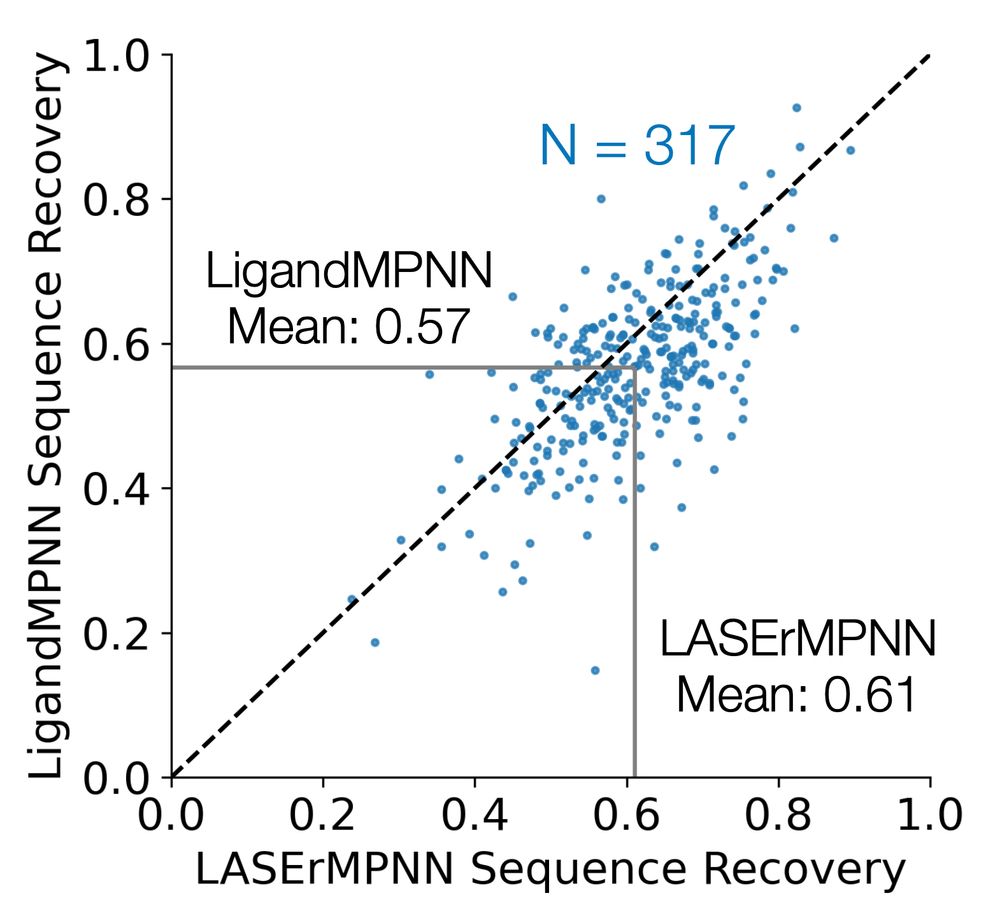
scatter plot of sequence recovery on a test set showing lasermpnn improves slightly over ligandmpnn
Ben Fry (@benf549.bsky.social) was excited when proteinMPNN came out, which motivated him to train a new gNN called LASErMPNN to design sequences given protein-ligand co-structure. LASErMPNN does pretty well at this! The repo is available and even has the training code! github.com/polizzilab/L...
28.04.2025 15:22 — 👍 3 🔁 1 💬 1 📌 0
Awesome to see a fully open source model. Looking forward to using this Gabriele!
18.11.2024 00:33 — 👍 1 🔁 0 💬 1 📌 0
chemical biology enthusiast & strategist; I make impossible things possible - all before breakfast!
I use robots and biophysics to study cancer therapies in the Chodera Lab at Memorial Sloan Kettering Cancer Center. Views are my own.
MSCA fellow at @crg.eu w/M. Dias and @jonnyfrazer.bsky.social
> Biological Physics | Proteins | Comp Bio | ML
https://scholar.google.com/citations?user=n55NtEsAAAAJ&hl=en
Evolutionary biochemist and protein complex enthusiast at the University of Marburg.
Harvard PhD Candidate SSQB
@DeboraMarksLab
Turtles all the way down
https://www.courtneyshearer.com/
Research in AI for Protein Design @Harvard | Prev. CS PhD @UniofOxford, Maths & Physics @Polytechnique
Associate Professor of Machine Learning, University of Oxford;
OATML Group Leader;
Director of Research at the UK government's AI Safety Institute (formerly UK Taskforce on Frontier AI)
PhD Candidate, UCSF Biophysics, Kortemme Lab
Computational Biology & AI Lead, Animate Bio
ML for Protein Design
Postdoc @ Debbie Marks Lab, Harvard | Prev. PhD @ MIT EECS || ML for Proteins + Viruses 🦠
Decoding biology to radically improve lives. The industrial revolution of drug discovery is here. Learn more at recursion.com #techbio
Drug discovery scientist at Recursion, previously Exscientia, molecular pharmacology PhD, Pac NW born and raised, ex-expat back in the USA, dog person, occasional sleeper. Opinions are my own and obviously correct.
Final year chemistry PhD candidate @ Boston Uni. Explores applications & method development for understanding molecular recognition, evaluating protein druggability, and elucidating the impact of protein dynamics in these aspects.
Full stack developer who occasionally wears a lab coat. Graduate student at Harvard Medical School.
Discover the Languages of Biology
Build computational models to (help) solve biology? Join us! https://www.deboramarkslab.com
DM or mail me!
Associate professor Duke BME
www.romerolab.org
Asst prof at HMS, PI at DFCI
Designing proteins
polizzilab.org
PhD student@MIT
https://sites.google.com/view/yehlincho/home