If you're otherwise at the conference and would like to meet up, shoot me a DM!
01.12.2025 22:23 — 👍 1 🔁 0 💬 0 📌 0
Excited about AI for Biology? At NeurIPS? Come say hi to the Biohub (née EvolutionaryScale) folks in the exhibit hall!
Keep me company:
Tue 12-3
Wed 3-6
Or stop by any time for a chance to meet my incredible colleagues: roshanrao.bsky.social, ebetica.bsky.social, rsmolina.bsky.social et al.
01.12.2025 22:23 — 👍 3 🔁 1 💬 1 📌 0
Rarely does a play make you apply to grad school. RIP
29.11.2025 18:07 — 👍 1 🔁 0 💬 0 📌 0

This October I’m drawing one molecule a day inspired by proteins @rcsb.bsky.social
Day 1/31
Prompt MUSTACHE
Pdb 2QZI
Let’s start with something fun:
Mr. Potato head’s ‘stache is made of Androgen Receptor that binds testosterone and helps maintain his male phenotype
Next prompt: WEAVE
suggestions?
02.10.2025 03:09 — 👍 28 🔁 8 💬 2 📌 0

We are excited to share GPN-Star, a cost-effective, biologically grounded genomic language modeling framework that achieves state-of-the-art performance across a wide range of variant effect prediction tasks relevant to human genetics.
www.biorxiv.org/content/10.1...
(1/n)
22.09.2025 05:29 — 👍 175 🔁 90 💬 4 📌 5
slides remain the hardest modality
22.08.2025 07:22 — 👍 5 🔁 0 💬 0 📌 0
https://authors.elsevier.com/a/1lbX08YyDfuZWX
Antibodies are highly diverse, but most possible sequences are unstable or polyreactive. In this work, just published in Cell Syst., we propose a new source of data for modeling constraints from these properties. Our models show clear improvements in predicting Ab dysfunction. (1/n)
t.co/qCZERPUMPF
15.08.2025 13:17 — 👍 16 🔁 6 💬 1 📌 0
Sign up for The Tournament!
🌍 Design a PETase - real-world impact on bioremediation!
🧬 Sponsored DNA synthesis and functional screening - no need for a lab!
🤖 Sponsored ESM inference through @evolutionaryscale.bsky.social Forge - if GPUs are a barrier!
🏆 Winners get published and win up to $15K!
13.08.2025 21:09 — 👍 2 🔁 0 💬 0 📌 0

Why PETase for our tournament? In 2024, the world made about 30 million tonnes of PET plastic, most from fossil fuels.
PETase can degrade PET, but isn’t ready for industrial-scale waste. The challenge: design an improved variant that can change that.
Register by Oct 17 alignbio.org/protein-engi...
13.08.2025 17:38 — 👍 7 🔁 2 💬 0 📌 1
With Tom Lehrer's passing, I suppose this is a moment to share the story of the prank he played on the National Security Agency, and how it went undiscovered for nearly 60 years.
27.07.2025 21:01 — 👍 8661 🔁 3616 💬 143 📌 717
Stats friends... what would your estimator be if you were interested in a similar question as this study that is lighting Bluesky on fire tonight? 1/x
11.07.2025 00:08 — 👍 21 🔁 1 💬 5 📌 1

1/4
🚀 Announcing the 2025 Protein Engineering Tournament.
This year’s challenge: design PETase enzymes, which degrade the type of plastic in bottles. Can AI-guided protein design help solve the climate crisis? Let’s find out! ⬇️
#AIforBiology #ClimateTech #ProteinEngineering #OpenScience
08.07.2025 16:26 — 👍 23 🔁 20 💬 1 📌 4
We're sponsoring the use of ESM3 and EMSC to help researchers engineer improved PETase enzymes in the @AlignBio 2025 Protein Engineering Tournament.
Get started using ESMC to predict protein function and ESM3 to generate new enzymes here: github.com/evolutionary...
08.07.2025 18:01 — 👍 9 🔁 3 💬 0 📌 1
Today I remembered my first QM parameterization of a small molecule failed miserably (turn volume ON for a full experience)
26.03.2025 21:18 — 👍 29 🔁 4 💬 3 📌 1
NIH funding supporting the HMMER and Infernal software projects has been terminated. NIH states that our work, as well as all other federally funded research at Harvard, is of no benefit to the US.
22.05.2025 12:42 — 👍 287 🔁 232 💬 37 📌 47

Thrilled to see my digital art on the cover of Trends Genet. The two binary strings represent reverse-complementary DNA sequences (00=A, 01=C, 10=G, 11=T) and the connecting rectangles represent “embeddings” learned by DNA language models. Pls check out our article as well: doi.org/10.1016/j.ti...
07.04.2025 15:01 — 👍 69 🔁 13 💬 0 📌 1
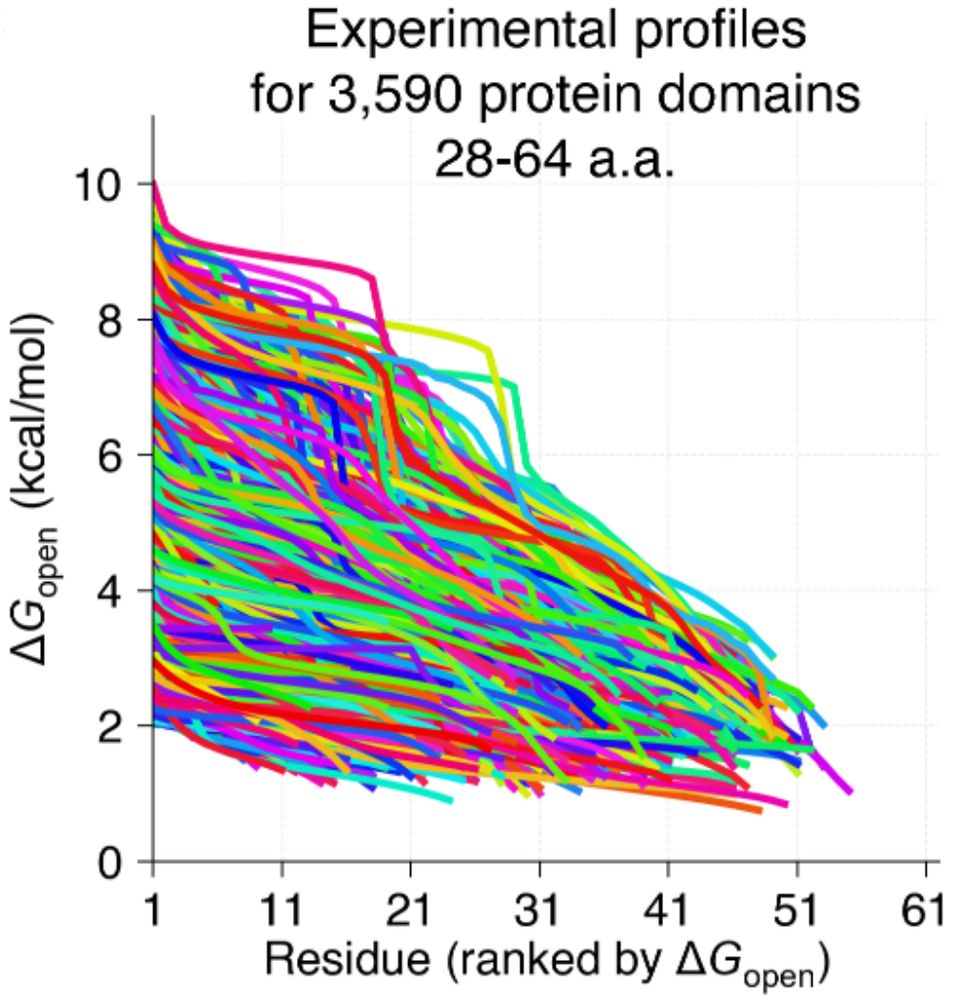
Small proteins can be more complex than they look!
We know proteins fluctuate between different conformations- but by how much? How does it vary from protein to protein? Can highly stable domains have low stability segments? @ajrferrari.bsky.social experimentally tested >5,000 domains to find out!
26.03.2025 16:21 — 👍 86 🔁 36 💬 4 📌 0
Gene synthesis is often the most expensive part of protein engineering with generative models.
Happy to have played a small part in this work, where Chase developed a method for precision library construction at scale, with per-gene costs as low as $1.50.
@philromero.bsky.social
24.03.2025 17:24 — 👍 63 🔁 23 💬 1 📌 0
So exciting to think what we will be able to do as we pair scaled library assembly techniques like these with ML-designed libraries and high throughput screening!
24.03.2025 17:38 — 👍 4 🔁 0 💬 0 📌 0
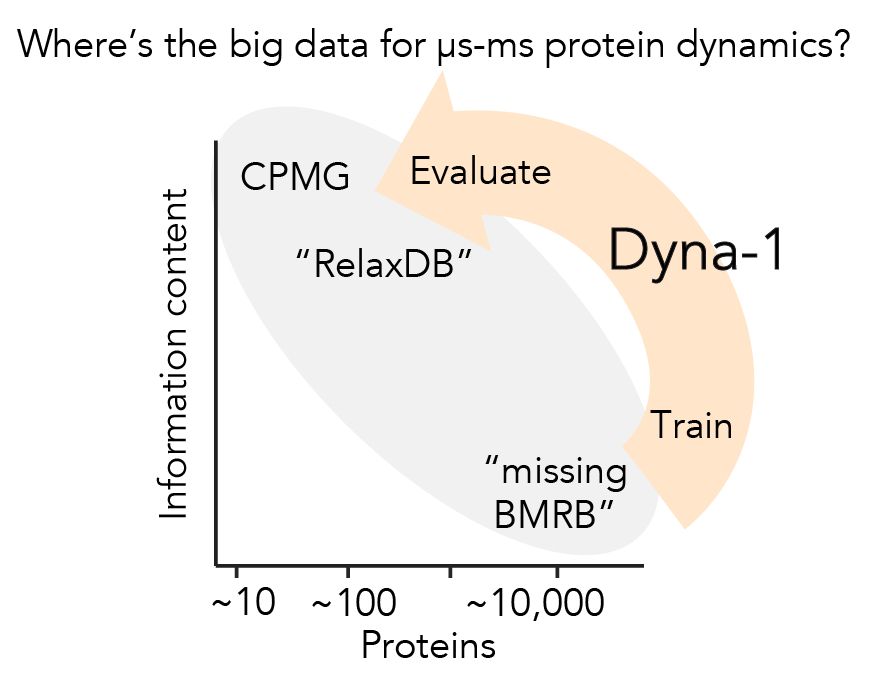
Protein dynamics was the first research to enchant me >10yrs ago, but I left in PhD bc I couldn't find big experimental data to evaluate models.
Today w @ginaelnesr.bsky.social, I'm thrilled to share the big dynamics data I've been dreaming of, and the mdl we trained w them: Dyna-1.
📝: rb.gy/de5axp
20.03.2025 15:02 — 👍 84 🔁 25 💬 2 📌 2
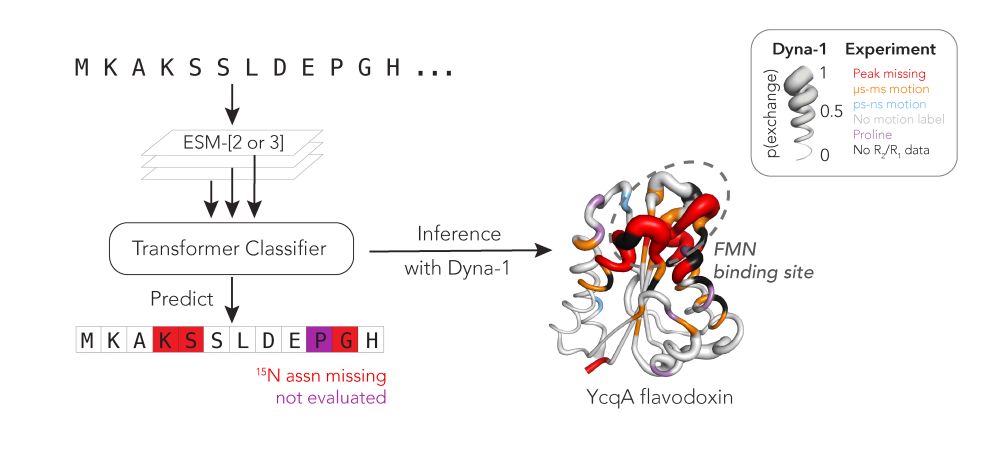
Protein function often depends on protein dynamics. To design proteins that function like natural ones, how do we predict their dynamics?
@hkws.bsky.social and I are thrilled to share the first big, experimental datasets on protein dynamics and our new model: Dyna-1!
🧵
20.03.2025 15:02 — 👍 103 🔁 38 💬 6 📌 5
YouTube video by ML for protein engineering seminar series
Engineering highly active and diverse nuclease enzymes by ML and high-throughput screening
If you’re interested in learning more, check out our @ml4proteins.bsky.social seminar talk on this work
www.youtube.com/watch?v=eGNE...
12.03.2025 17:18 — 👍 1 🔁 0 💬 1 📌 0
This was a collaborative effort between myself, David Belanger, Lucy Colwell and our whole team: Chenling Xu, Hanson Lee, Kathleen Hirano, Kosuke Iwai, Vanja Polic, Kendra Nyberg, Kevin Hoff, Lucas Frenz, Charlie Emrich, Jun Kim, Mariya Chavarha, Abi Ramanan, Jeremy Agresti
12.03.2025 17:18 — 👍 0 🔁 0 💬 1 📌 0
This campaign was completed in 2021! Since then, the field has evolved tremendously. We’re excited about work that pushes forward:
1) Multi-objective optimization
2) Generative models (e.g. ESM3, ProGen, RFDiffusion)
3) Synergy with randomized library design
… to name a few
12.03.2025 17:18 — 👍 1 🔁 0 💬 1 📌 0
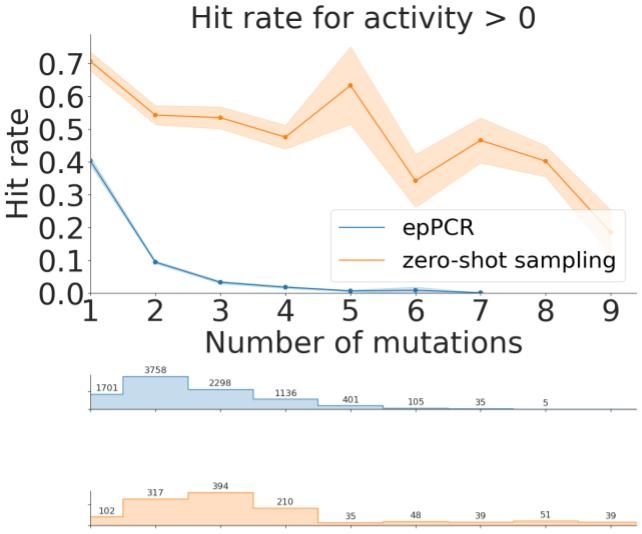
Multiple Sequence Alignments (MSAs) were also powerful for zero-shot design! Without any assay data, and even without structure or large-scale pretraining, we were able to design improved NucB variants with as many as 9 mutations from the wildtype.
12.03.2025 17:18 — 👍 1 🔁 0 💬 1 📌 0
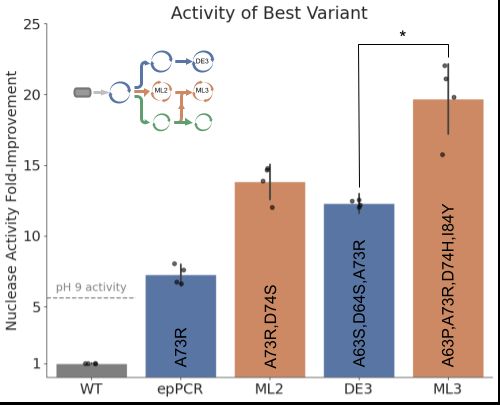
We found that in a head-to-head comparison of ML-guided design versus high-throughput directed evolution, our ML system could design higher activity variants, with lots more diversity!
12.03.2025 17:18 — 👍 1 🔁 0 💬 1 📌 0
MIT biology lab using computational and experimental methods to study protein structure, function, and interactions
I work at @letta.com.
We build infrastructure for self-improving artificial intelligence.
An element of truth - videos about science, education, and interesting things.
🌏🌦️🌊♻️🚲🥏🛹🏄♀️🏋️🏃♀️⚽️🥐💙💛🐻
Postdoc at Penn Genetics working in statistical genomics and computational biology.
Anti-cynic. Towards a weirder future. Reinforcement Learning, Autonomous Vehicles, transportation systems, the works. Asst. Prof at NYU
https://emerge-lab.github.io
https://www.admonymous.co/eugenevinitsky
I ♥ evolution, immunology, math, & computers. Professor at Fred Hutch & Investigator at HHMI. http://matsen.fredhutch.org/
The Complete Work of Charles Darwin Online (https://darwin-online.org.uk/)
News from Jennifer Doudna's lab at UC Berkeley, Innovative Genomics Institute. Tweets from lab members and not Jennifer Doudna unless signed JD. Tweets represent personal views only.
doudnalab.org
A LLN - large language Nathan - (RL, RLHF, society, robotics), athlete, yogi, chef
Writes http://interconnects.ai
At Ai2 via HuggingFace, Berkeley, and normal places
Postdoc @ Princeton AI Lab
Natural and Artificial Minds
Prev: PhD @ Brown, MIT FutureTech
Website: https://annatsv.github.io/
Ribosomes and RNA language models.
Professor at UC Berkeley.
https://sites.google.com/berkeley.edu/cate-lab/home
Pushing flies at UC Berkeley | Functional evolutionary geneticist, interested in toxins/insects/microbes
Combining frontier AI & frontier biology to help scientists cure or prevent disease
Images, AI, ML, and ALife.
Former: @hampshirecolg
@brandeisuniversity.bsky.social
@harvardmed.bsky.social
@uidaho.bsky.social
@hhmijanelia.bsky.social
@mdc-berlin.bsky.social
@ORNL.
Now @biohub.org.
Opinions are mine.
Mol bio | Genomics | Development | AI
PhD @Radboud uni, postdoc @ EMBL, Genomics scientist @Google DeepMind
Fragile Nucleosome co-organiser
Views are my own
Associate professor at ETH Zurich, studying the cellular consequences of genetic variation. Affiliated with the Swiss Institute of Bioinformatics and a part of the LOOP Zurich.