We are hiring for a metabolomics position here at UC Riverside! Come join the vibrant research and mass spectrometry community here!
aprecruit.ucr.edu/JPF02151

@mingxunwang.bsky.social
Assistant Professor @ UCR Computational Mass Spectrometry, Bioinformatics. #massspec #molecularnetworking #GNPS #MassQL https://www.cs.ucr.edu/~mingxunw/
We are hiring for a metabolomics position here at UC Riverside! Come join the vibrant research and mass spectrometry community here!
aprecruit.ucr.edu/JPF02151

To help make more mass spec data accessible - we've just rolled out a change to enable universal spectrum identifier resolution and plotting directly from mzML files in Zenodo. We're growing support from more sources in GNPS2 for public data reanalysis!
metabolomics-usi.gnps2.org/dashinterfac...

Interested in a co-authorship?
We’re building a tool for repository-scale untargeted #metabolomics and #exposomics of #environmental data. To make it the best it can be, we’re looking for people willing to share high-resolution LC-MS/MS (DDA) data from #water, #soil, #sediment, and related samples.
GNPS2 and associated services will be down for power maintenance tonight and into tomorrow.
02.08.2025 04:48 — 👍 2 🔁 1 💬 0 📌 0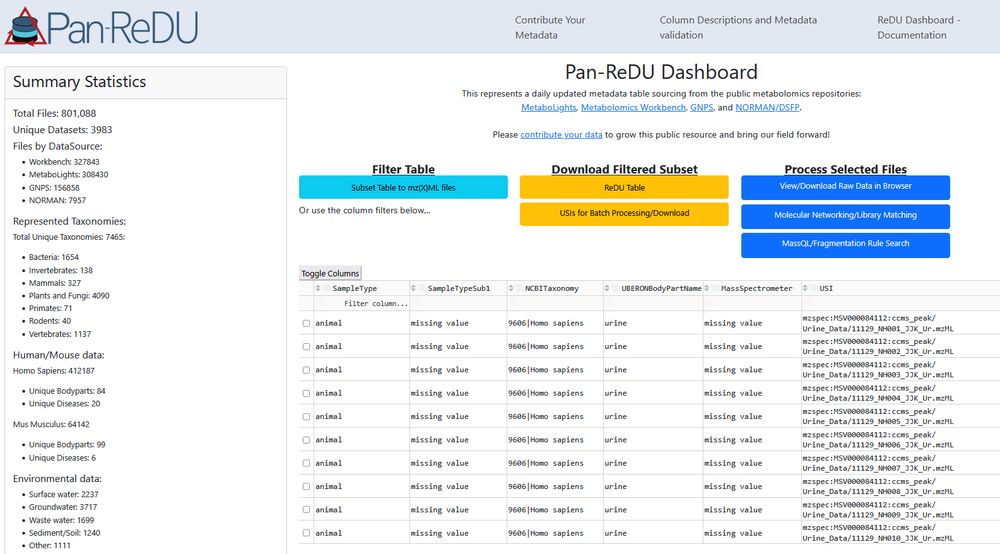
We just crossed the 800,000 files mark in Pan-ReDU. That's 800,000 public #metabolomics raw data files with harmonized metadata that can be re-analyzed to learn about new molecules and bio-distributions. 🎉 redu.gnps2.org
09.07.2025 00:55 — 👍 16 🔁 6 💬 0 📌 0still in development
26.06.2025 18:47 — 👍 1 🔁 0 💬 0 📌 0Yes, don't use that for the moment in classical networking
26.06.2025 18:47 — 👍 1 🔁 0 💬 1 📌 0Yes, you can do that, thats the default intensity threshold. That is relative to the base peak in the MS2.
18.06.2025 20:32 — 👍 1 🔁 0 💬 1 📌 0So now the 85 peak will need to be within 10% of the intensity of the 393 peak. You can put any expression on the 85 peak to modulate up or down for what you want
18.06.2025 16:04 — 👍 0 🔁 0 💬 1 📌 0QUERY scaninfo(MS2DATA) WHERE MS2PREC=393.2283:TOLERANCEMZ=0.1: INTENSITYMATCH=Y:INTENSITYMATCHREFERENCE AND MS2PROD=85.029:TOLERANCEMZ=0.1: INTENSITYMATCH=Y:INTENSITYPERCENT=10
18.06.2025 16:04 — 👍 1 🔁 0 💬 2 📌 0Hi @galanojeanmarie.bsky.social Yes, you're just missing one thing with the variable Y to determine the peak intensity of the second one.
18.06.2025 16:04 — 👍 1 🔁 0 💬 2 📌 0LOL - fun problem to have. I think this might be possible - I think the main graphml, we'll just need to get the actual task and display title.
28.05.2025 03:35 — 👍 2 🔁 0 💬 0 📌 0Thanks for the feedback - let me see if we can integrate. We've already added direct links for modifinder - so we can easily push it out to the resolver as well with the mirror plots.
28.05.2025 03:35 — 👍 1 🔁 0 💬 0 📌 0
The Mass Spectrometry Query Language (MassQL) is an open-source language for instrument-independent searching across mass spectrometry data for complex patterns of interest via concise and expressive queries without the need for programming skills.
www.nature.com/articles/s41...
Thanks @ucriverside.bsky.social for featuring our work!
news.ucr.edu/articles/202...

If you're new to MassQL - definitely checkout the research briefing that describes how MassQL enables scientists to precisely express and reproducibly search for mass spectrometry patterns in large datasets:
www.nature.com/articles/s41...
and find the full article:
www.nature.com/articles/s41...
I am thrilled to share after years of work/procrastination that the MassQL manuscript is finally published in @natmethods.nature.com - "A universal language for finding mass spectrometry data patterns". This was an team effort from all co-authors that helped shape MassQL and how it could be used.
12.05.2025 18:10 — 👍 40 🔁 17 💬 1 📌 2We are back online!
06.03.2025 04:42 — 👍 8 🔁 2 💬 0 📌 0
MS-RT: A Method for Evaluating MS/MS Clustering Performance for Metabolomics Data pubs.acs.org/doi/10.1021/...
06.03.2025 20:34 — 👍 6 🔁 2 💬 0 📌 0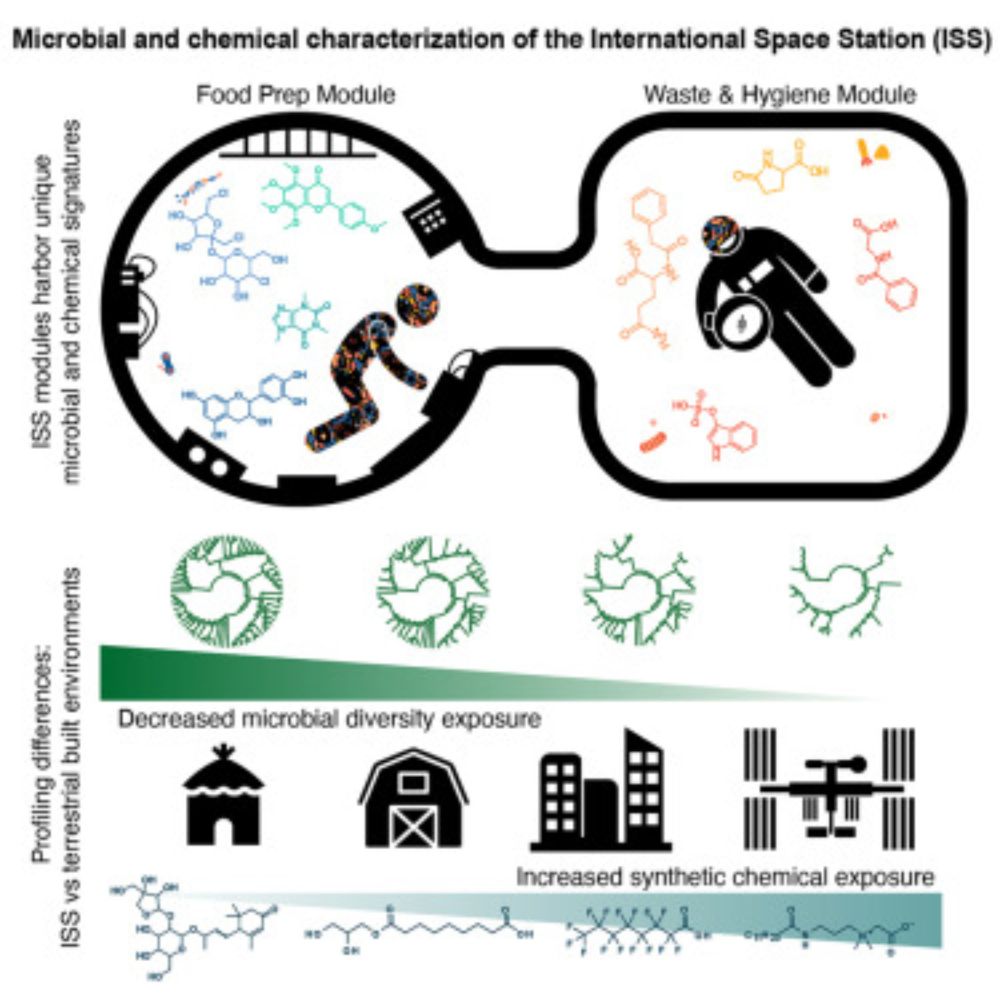
Congrats to all the people who put in tremendous effort to make this study possible. Such a fun project!
www.cell.com/cell/fulltex...
#space #metabolomics #microbes

#FeaturedProtocol this week is a #reversemetabolomics protocol, in which a tandem #massspec spectrum is used as a search term to probe public #metabolomic data, enabling discovery of new metabolic associations bit.ly/4hdyQQF
04.03.2025 15:21 — 👍 8 🔁 3 💬 0 📌 0Big thanks to Xianghu Wang for all the work as lead author and all coauthors who made this possible and the funding from @corteva.bsky.social
05.03.2025 17:34 — 👍 2 🔁 0 💬 0 📌 0While most of the clustering innovation in mass spectrometry has focused largely in proteomics - we hypothesize due to the ability to assess performance - I hope that tools like MS-RT can accelerate the computational innovation in metabolomics.
05.03.2025 17:34 — 👍 1 🔁 0 💬 1 📌 0
After validation, we used MS-RT to evaluate the performance of several commonly used MS/MS clustering tools used in proteomics, specifically MS-Cluster and Falcon. We found that Falcon made generally favorable tradeoffs between purity can clustering completeness (how much was actually clustered).
05.03.2025 17:34 — 👍 0 🔁 0 💬 1 📌 0We validate this MS-RT approach by using proteomics MS/MS dataset and comparing the purity estimates from MS-RT with estimates using state-of-the-art proteomics database search approaches. We found that while not exactly identical the relative order across clustering tools is maintained.
05.03.2025 17:34 — 👍 1 🔁 0 💬 1 📌 0To address this, we introduce MS-RT which uses the retention time dimension within individual LC/MS/MS dataset to estimate the clustering purity (how often different molecules make it into the same MS/MS cluster).
05.03.2025 17:34 — 👍 2 🔁 0 💬 1 📌 0While we're accustomed to being able to evaluate clustering performance in MS/MS proteomics clustering - by using FDR controlled database search - this is a missing piece in MS/MS clustering in metabolomics.
05.03.2025 17:34 — 👍 1 🔁 0 💬 1 📌 0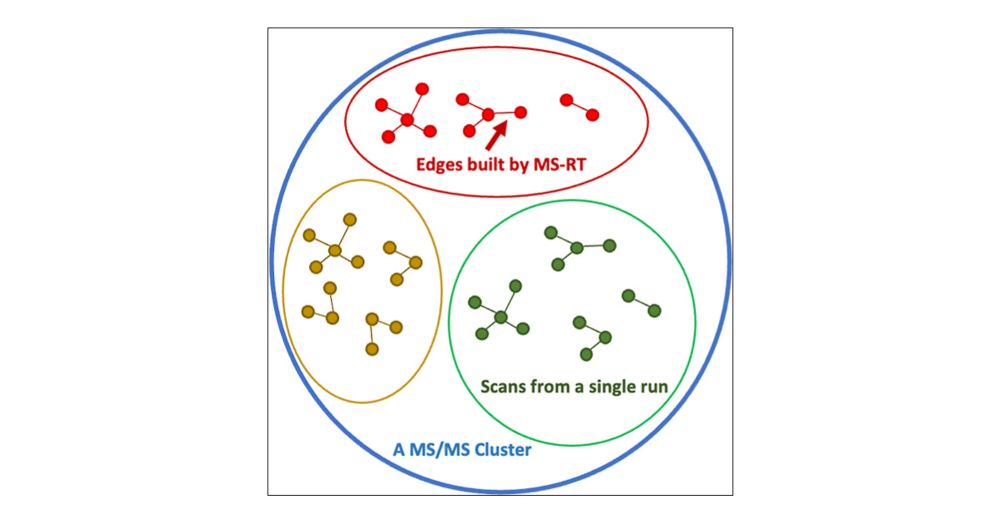
I am excited to share this new paper out in JPR - "MS-RT: A Method for Evaluating MS/MS Clustering Performance for Metabolomics Data." This work introduces the MS-RT method to assess MS/MS clustering accuracy on metabolomics data.
doi.org/10.1021/acs....
GNPS2 is planning on being down for server maintenance tomorrow at 12PM PST. We expect 5 hours of downtime to move servers, bring online new storage, and increase networking performance.
05.03.2025 03:45 — 👍 8 🔁 5 💬 0 📌 1Massive has a workflow that does this, I think called protein explorer
31.01.2025 23:22 — 👍 1 🔁 0 💬 1 📌 0